早在1959年《藥物化學雜志》創刊伊始,便有了乙炔(乙炔基)在藥物化學中應用的記載。此后,乙炔基在藥物的發現和開發中得到了廣泛的應用。在藥物活性中發揮多種作用,其中包括效力增強(通過與受體結合口袋的互補配合)、反應彈頭(不可逆地抑制目標蛋白) 、非極性線性剛性間隔(使藥效團附屬物以有利的幾何形狀進行組裝)、眾多官能團的生物等排體(即氰、氯、碘、酰胺、乙烯、羰基、乙基、苯基和環丙基)以及藥物代謝動力學(DMPK)的調節。此外,化學生物學探針中經常引入端炔基團,作為識別分子靶點以及評估目標靶點參與情況的可點擊手柄。然而,對非目標分子CYP450/GSH等的共價抑制也使得乙炔基團也具有一定的毒副作用。發展至今,乙炔基團的已知特性如下圖所示: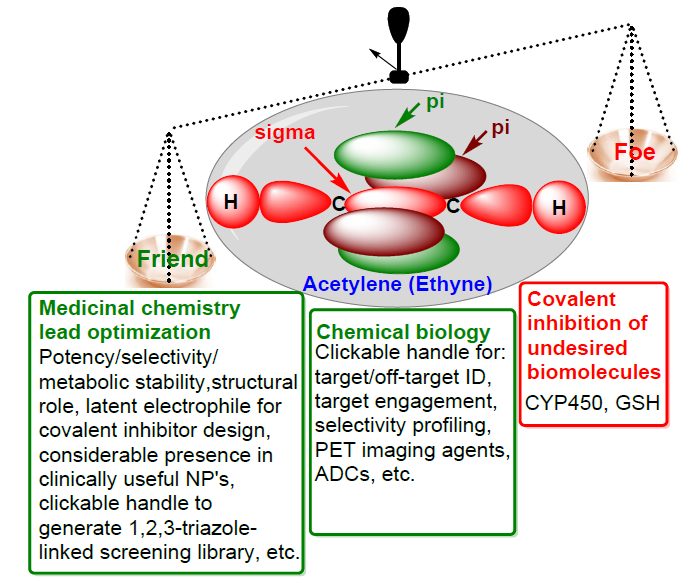
圖1 乙炔基團的特性
乙炔,由埃德蒙·戴維于1836年發現,是一種無色無味的氣體。它由一個碳碳三鍵組成,其中具有一個σ鍵和兩個π鍵。乙炔的三鍵較短(1.21A),但穩定性強于碳碳雙鍵(1.34A)或單鍵(1.53A)。例如,乙炔的π鍵斷裂需要318kJ/mol,而乙烯π鍵斷裂則只需要268kJ/mol。由于其S軌道的增加,乙炔C-H鍵(1.06A)比相應乙烯(1.08A)或乙烷(1.09A)的C-H鍵短。當它與烷基基團相連時,乙炔基團是通過超共軛(π鍵與相鄰的C-H鍵重疊)來實現穩定的。因此,內炔比端炔更穩定。由于乙炔是一個弱吸電子基團,它可以作為質子的供體。因此與相應的烯烴(pKa~44)或烷烴(pKa~50)相比,端炔是更強的Bronsted酸(pKa~25)。乙炔基的H-C-C鍵角是線性的(夾角呈180度),因此它可以作為一個剛性的間隔,使得取代基處于乙炔基的同一平面上。乙炔基團π鍵體系提供的供體-受體相互作用與在苯環中觀察到的類似,因此,它也可以作為苯基的電子等排體。
雖然乙炔是一種碳氫化合物,但它被認為是一個官能團,因為它的C-H鍵可以在堿性條件下去質子化,產生共軛堿。同樣地,它可以在親電π鍵上進行親核加成。因此,乙炔基團在許多基團的合成中占有重要地位,包括烷烴、烯烴、醛、鹵化物、雜環和閉合環分解等反應。銅催化的疊氮和炔基的[3+2]環加成(CuAAC)反應,可迅速生成1,4-二取代的1,2,3-三唑,是一種典型的“點擊化學反應”,該反應在醫藥化學中被廣泛應用于先導物有效優化。
基于乙炔基的電子排布特點,乙炔基團被看做是一種非典型的生物等排體。在生物體內可進行等效替換的官能團有氯、氰、碘、乙烯、乙基、羰基、羧胺、苯和環丙基等基團。在乙炔、乙烯和苯基中存在相似的π鍵體系和供-受體相互作用,使它們成為彼此的生物電子等排體。它的π電子云與芳香環的非常類似,其極化的-CH鍵有弱的氫鍵供體作用,并可以被碘原子取代。此外,氯、溴和碘取代的苯的分子靜電勢與苯乙炔的分子靜電勢相似,乙炔基和苯之間的鍵長與碘原子與苯的鍵長相當,而且乙炔的弱氫鍵能力與鹵素鍵的強度相當,通過Sonogashira偶聯反應,芳香環上碘原子可輕易被乙炔基等電置換。由此可見乙炔為鹵素的生物電子等排體。氰基和乙炔基團在結構上有相似之處,均存在sp雜化中心,空間上呈現短的極化的線性三鍵。進一步研究表明,乙炔中的sp雜化末端碳的電負性為3.29,而氰基中的sp雜化氮的電負性為5.07,表明兩者都是吸電子基團,并且氰基比乙炔的吸電子能力更強。因此,乙炔和氰基可互為生物等排體。此外,乙炔基團還可以功能性的模擬酰胺、羰基、乙基和環丙基等基團,這可以通過本文提供的例子得到驗證。
以Mtb(結核分枝桿菌)中PknA/PknB雙抑制劑1和2為例,具體闡述乙炔基作為生物等排體在藥物化學中的應用。結核分枝桿菌的蛋白激酶A(PknA)和蛋白激酶B(PknB)由于其在細菌細胞生長中的重要作用,藥物研發時以該兩種激酶為靶點,以獲得有效的抑制劑,從而降低結核分枝桿菌耐藥性。如圖2所示,研究表明,化合物1對兩種激酶具有微摩爾級別的抑制作用。但對哺乳動物細胞周期蛋白依賴性蛋白激酶CDK2的選擇性較差。1與PknA和PknB結合的X射線晶體結構顯示,甘氨酸殘基翻轉形成的口袋(Gly100 in PknA and Gly97 in PknB)導致化合物1對哺乳動物細胞周期蛋白依賴性蛋白激酶CDK2的選擇性較差,而對Mtb激酶的選擇性較好。化合物2中是用5-乙炔基取代化合物1中的5-氯基時,不僅保持了化合物2對Mtb激酶的效力以及抗菌活性,還提高了對CDK2激酶的選擇性。由化合物1與2的抑菌效力可見,炔基與鹵素氯互為生物等排體。
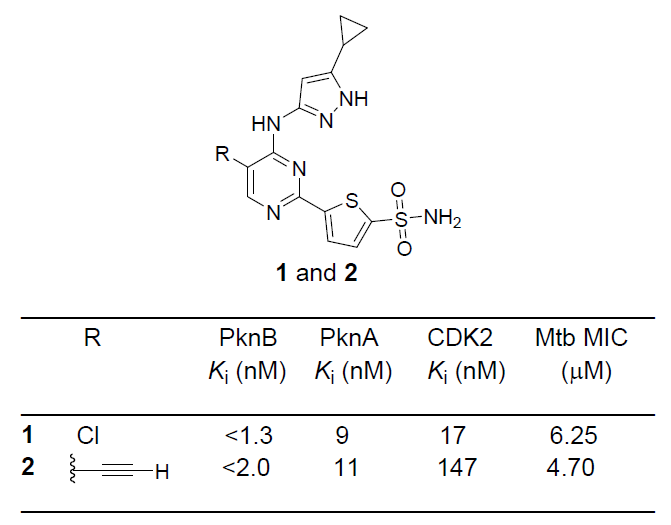
圖2 化合物1、2的結構及其在體內的相關性質
Dragovich和他的同事報告了人類鼻病毒3C蛋白酶2-吡啶酮類抑制劑的藥代動力學優化過程。如圖3所示,化合物3中存在的3,4-二氟芐基部分被一個炔丙基所取代以獲得化合物4,雖然效力有所下降。但化合物4的極性、水溶性、代謝穩定性增加,血漿半衰期延長,藥代動力學性質顯著改善。
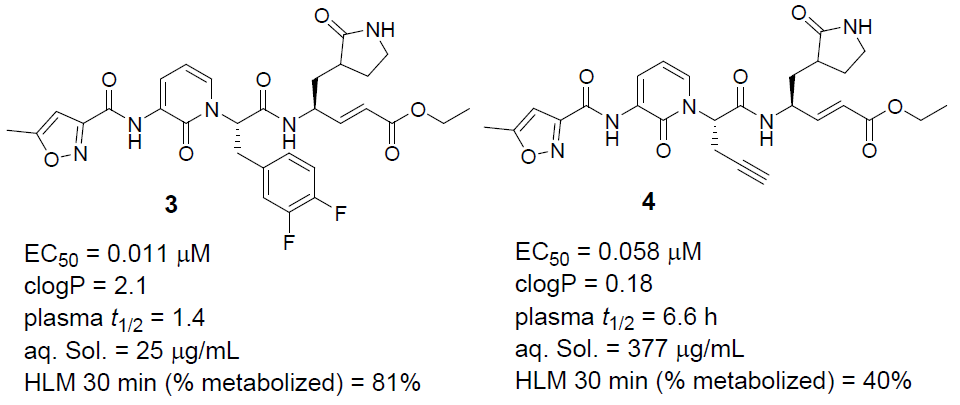
圖3 人鼻病毒3C蛋白酶抑制劑3(圖左)和4(圖右)的結構及其體外效力和DMPK數據
開發具有明確靶向性的抑制劑一直是藥物學家限制脫靶效應的首要任務。為了實現這一目標,可在分子內插入乙炔基團以提高分子靶向性。DPP-4抑制劑即二肽基肽酶 Ⅳ抑制劑,是治療2型糖尿病的新靶點, 能夠迅速滅活腸促胰島素胰高血糖素樣肽-1( GLP-1)和糖依賴性胰島素釋放肽(GIP)等多種激素。提高內源性GLP-1和GIP的活性,促進胰島B細胞釋放胰島素,同時抑制胰島 A 細胞分泌胰高血糖素,從而提高胰島素水平,降低血糖,且不會誘發低血糖和增加體重。在二肽基肽酶 Ⅳ抑制劑(DPP-4)抑制劑中插入乙炔基團增強其靶向性成為DPP-4特異性抑制劑,對與其密切相關的DPP-8和DPP9亞型則不具備抑制性。如圖4所示,在化合物5的2-氰基吡咯烷部分的C5位置安裝了一個乙炔基團,得到化合物6,與5相比,6對DPP-4選擇性與DPP-8和DPP-9相比有相當大的提高。在吡咯環上保留氰基和乙炔取代基的基礎上,用C4-甲基哌啶取代了環己基羥基連接劑,并用異煙酸替換氰吡啶環獲得化合物7(ABT-279)。 進一步提高了對DPP-4的抑制效力和選擇性,對特異性治療2型糖尿病藥物的研發提供新途徑。
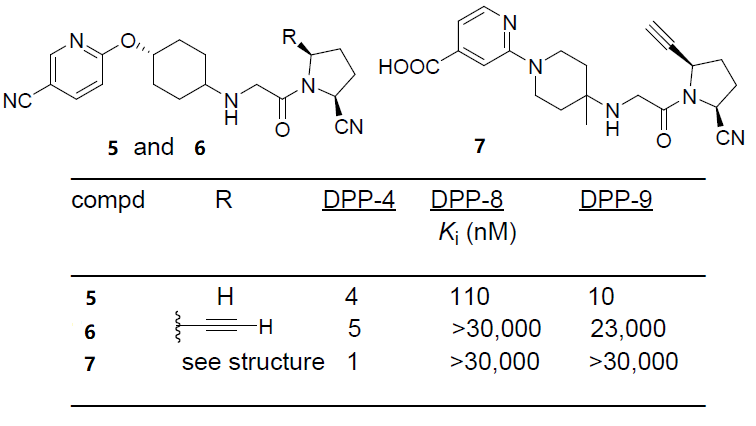
圖4 DPP-4抑制劑5-7的結構和體外效力
炔基由于其狹小的棒狀結構而被廣泛用作化合物內部的結構基序,以穿過靶蛋白內的空間限制通道。作為富電子基團,可增強與活性位點芳香殘基的π-π相互作用。它還是一種對空間要求較低的高效連接劑,可創造出與目標大分子上兩個相鄰結合位點特異性結合的對稱二聚體化合物。此外,它還可作為連接基團限制化合物的空間構象以及提高目標選擇性。以Bcr-Abl激酶抑制劑的發現 揭示了乙炔在化合物結構中發揮的作用。
BCR-Abl激酶在慢性粒細胞白血病(CML)中起著關鍵作用,因此,它是腫瘤學的重要靶點。如圖5所示,化合物8(伊馬替尼)在CML患者中的臨床耐藥性與BCR-Abl激酶中保守殘基Thr315Ile的突變有關。Ile315的側鏈變異膨脹后,與藥物伊馬替尼結合時產生空間碰撞,導致結合親和力喪失,從而產生耐藥性。在伊馬替尼結構中的腺嘌呤結合部分和變構口袋結合部分之間用乙炔做連接基產生的化合物9(帕納替尼),有效的避免了與BCR-Abl激酶中Ile315龐大的側鏈發生空間沖突。但帕納替尼也具有血塊和血管狹窄等副作用。為此,藥物學家使用一個新的腺嘌呤結合部分和變構口袋結合部分通過炔基連接產生了化合物10。化合物10對野生型以及Thr315Ile突變型BCR-Abl激酶均具有良好的抑制性。
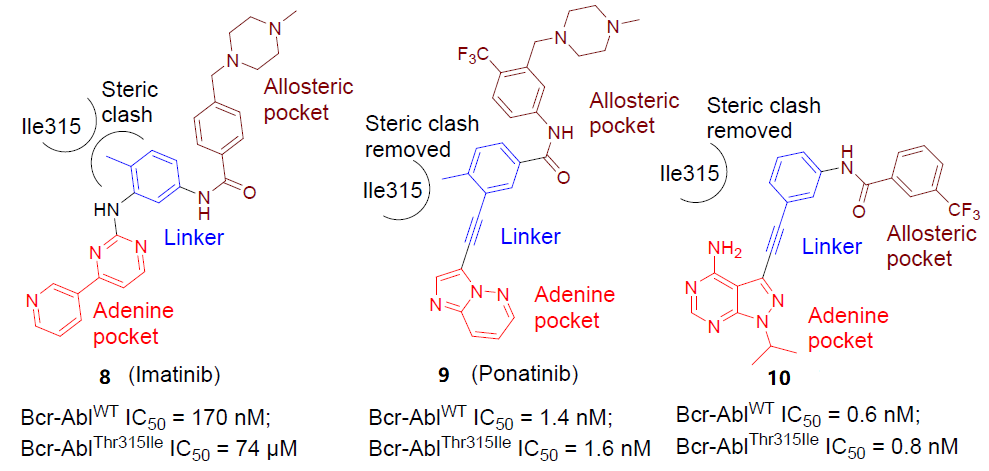
圖5 8-10的結構及相應Bcr-Abl激酶抑制活性
針對特定目標蛋白的高選擇性共價抑制劑在臨床上的應用越來越廣泛,然而,抑制劑中使用高活性的親電彈頭有可能導致對非目標蛋白不加區分的共價修飾并產生毒性。因此,使用反應活性較低的親電彈頭基團,如炔基,則成為不可逆親電抑制劑研發的重點方向。以將組織蛋白酶K(CatK)的可逆抑制劑11(Odanacatib)轉化為不可逆的潛在親電抑制劑12為例,闡述乙炔在不可逆抑制劑中作為潛在親電基的應用。
在破骨細胞Cat K中過表達的半胱氨酰蛋白酶參與了骨降解過程。因此,CatK的小分子抑制劑對于治療骨質疏松癥是有用的。化合物11(odanacatib)由于存在與半胱氨酸具有反應性的腈基,是一種高選擇性的Cat K可逆共價抑制劑。由于不可逆抑制劑中的高活性親電基團伴有的特定毒性,Mons和同事開發了含有低活性彈頭的CatK不可逆抑制劑,如N-proparylamide。研究人員用炔基取代了11中的腈基,卻發現化合物水溶性降低,這一問題通過去除疏水螺環丙基部分解決,產生化合物12。與已知的丙炔酰胺彈頭相比,12中N-丙炔酰胺彈頭表現出較低的親電性。化合物12中丙炔酰胺鍵通過與CatK中半胱氨酸的硫原子形成不可逆硫醚鍵從而發揮抗骨質疏松的作用。12對CatK的不可逆抑制作用機制如下圖所示:
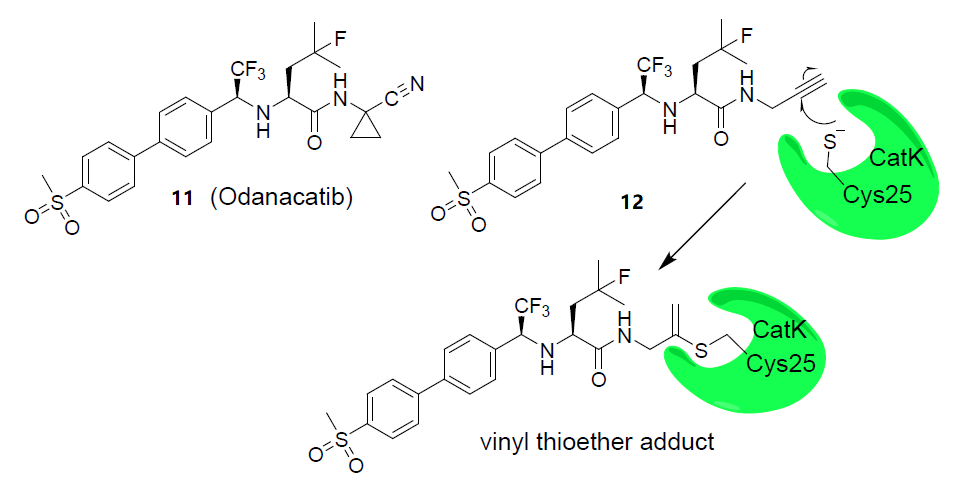
圖6 蛋白酶K(CatK)可逆共價抑制劑11和不可逆共價抑制劑12的結構及其作用機制
配體分子大分子水平機理的確定,包括目標受體和非目標受體的識別,已成為化學生物學中研究的關鍵領域。插入分子手柄,如炔基或疊氮,通過熒光讀出或親和純化后進行質譜分析,可以檢測細胞蛋白質組中的目標結合蛋白。因此,銅-(I)催化疊氮-炔[3+2]環加成反應(CuAAC)在許多化學生物學研究中起著關鍵作用,使環肽的產生和抗體治療學的發展成為可能。含炔探針在生物化學領域的應用十分廣泛:用于標記埃博拉病毒感染的目標蛋白、青蒿素檢測、CYP450共價標記、活性蛋白分析(ABPP)、跟蹤萬古霉素誘導的細胞壁變化等。
抗萬古霉素腸球菌(VRE)是公共衛生的難題。萬古霉素的抗菌作用機制為抑制存在于真細菌中的革蘭氏陽性菌和革蘭氏陰性菌的細胞壁中肽聚糖(PG)生物合成。PG頂端為五肽鏈組成的雙糖,該五肽具有末端D-Ala-D-Ala肽序列。VRE細菌細胞通過合成頂端為D-Ala-D-乳酸的PG前體,進一步合成PG,因此,VRE細菌細胞可以在萬古霉素的壓力下生長和增殖。為了解PG可塑性結構與VRE細胞的關系,皮吉恩和他的同事設計并研究了化學探針17(如圖7),一種含有端炔基的抗萬古霉素腸球菌底物D-乳酸的類似物。該探針的特點是含有一個適合CuAAC點擊化學的炔基“化學手柄”,VanB連接酶將利用這種改性底物產生D-Ala-D-laclick PG-錨,隨后,用疊氮熒光團如6-羧基熒光素疊氮化物(6-FAM-azide)與炔基手柄進行CuAAC點擊化學反應。通過觀察熒光水平的增加,表明萬古霉素腸球菌耐藥的過程(如圖7)。皮吉恩和他的同事假設,模擬D-Ala-D-Ala二肽,合成D,D-二肽酶(VanX)底物類似物如18,將允許在體內詢問VRE細菌細胞中的Van X活性。二肽18具有與6-FAM-zaide進行點擊化學反應的炔基探針。這種化學探針熒光的減少,表明萬古霉素腸球菌的耐藥性降低。
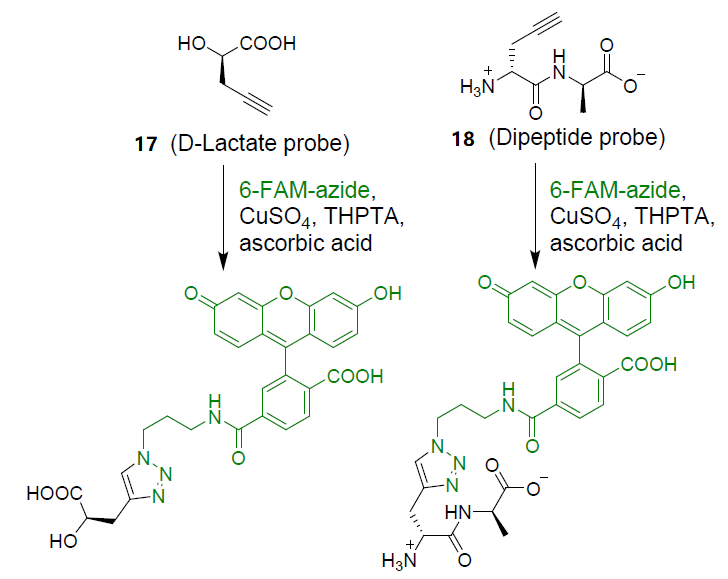
圖7 含炔探針17(D-乳酸)和18(二肽)結構及相應作用機制
乙炔基作為動力學靶向合成(KTGS)的點擊反應“手柄”
KTGS是利用原位點擊反應可生成的分子來抑制靶點 大分子(蛋白和DNA)作用,為先導物質的鑒定和優化提供了一種很有前途的策略。在KTGS中,一對具有互補反應性的高度親和力的片段與目標受體結合,受體加速高親和力片段間共價鍵形成,通過穩定互補片段與受體形成的三元配合物產物構象,使互補反應性的片段之間的反應不可逆,從而對目標受體自身產生抑制作用。(如圖8A所示)Antti和同事用碳酸酐酶II抑制劑進行了以細胞為基礎的KTGS研究。例如,將具有豐富的碳酸酐酶II的牛血紅細胞在疊氮化物19和含炔化合物20中培養,以實現化合物21的靶向合成(如圖8B所示)。疊氮化物19與含炔化合物20是具有高度親和力的互補片段,分別與RBCs/bCAII相應位點結合后,在RBCs/bCAII中發生點擊化學反應下形成共價鍵,生成化合物21。21與RBCs/bCAII的穩定結合使RBCs/bCAII的功能得到抑制。
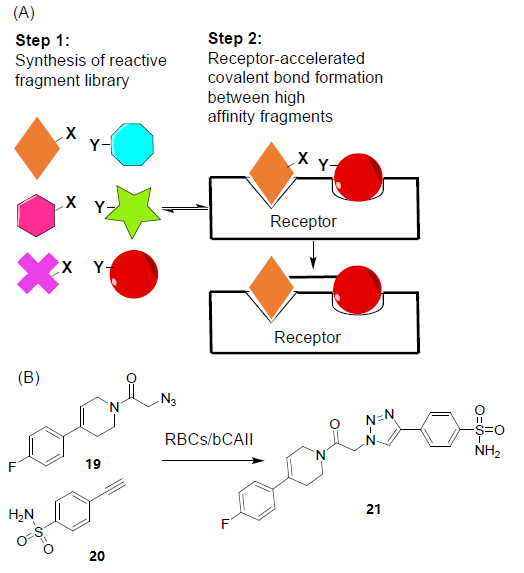
圖8 A靶向合成先導化合物示意圖 B以細胞為基礎的KTGS
乙炔基作為抗體-藥物偶聯物(ADC)開發的點擊反應“手柄”
抗體-藥物偶聯物(ADC)是一種通過化學連接物將高細胞毒性小分子(有效載荷)與人源化或人源性單克隆抗體連接產生的結合物,是一種新型的治療藥物,在癌癥化療中具有巨大的潛力。如圖9,格雷格森和他的同事報告了吡咯苯并二氮卓類藥物連接劑的設計,發展了使用關鍵中間體22的ADC。關鍵中間體22通過 Sonogashira反應以乙炔基取代碘原子生成化合物23,或通過對22進行乙炔基取代,隨后與適當功能化疊氮基發生點擊化學反應生成化合物24。23或24的馬來酰亞胺部分與抗HER2/抗CD22抗體的半胱氨酸結合形成ADC。這些ADC是由馬來酰亞胺基與HER2/ CD22抗體的半胱氨酸結合形成偶聯物、一個親水間隔、一個炔(23)或者一個1,4取代的1,2,3-三唑(24)與吡咯基苯二氮雜卓相連構成。

圖9 吡咯-苯二氮卓二聚體22的結構及相應含炔/三唑基的抗半胱氨酸HER2或抗CD22抗體23、24結構
由于乙炔在共價抑制劑設計和化學生物學研究中的應用,臨床上有用的含炔分子數量正在上升。盡管偶有位于分子內部和末端的乙炔對CYP酶有抑制的傾向,但它們已經在藥物化學中產生了許多有意義的貢獻。且合成乙炔的原料便宜易得,路線簡易,考慮到它們在化學生物學上的用途,預計乙炔在藥物化學中的應用會越發廣泛。
參考文獻:
[1] Acetylene Group, Friend or Foe in Medicinal Chemistry,Tanaji T. Talel,Journalof Medicinal Chemistry ArticleASAP,DOI:10.1021/acs.jmedchem.9b01617
注:本文圖片來源于參考文獻