酶工程廣泛應用于綠色生產工藝、診斷技術、治療應用和生物醫學等領域。定向進化和計算驅動的突變技術,通過調節底物專一性和提高酶的活性和穩定性來優化生物催化劑,促進酶工程的發展。在非催化蛋白支架中引入生物催化位點為此技術提供了新的機會。利用此技術構建的單活性位點人工酶催化的反應,其轉化率可媲美天然酶。其中,人工金屬酶(ArMs)的構建最引人注目,通過將催化活性強的有機金屬絡合物引入蛋白質中得到ArMs支架。
| |
|
| Wilson和Whitesides將二磷酸銠生物素衍生物引入鏈霉親和素支架。該非手性化學催化劑可用于α-乙酰氨基丙烯酸的手性加氫。然而,這項工作可能是由于催化性能令人失望,沒有引起科學界的注意。(JACS DOI: 10.1021/ja00469a064) |
| |
|
| 由于有機金屬催化和蛋白質工程的發展,產生了大量的人工金屬酶。盡管這類人工金屬酶可催化水解、還原、氧化以及C–C和C–雜原子等反應,但它們的催化性能一直不如天然金屬酶。(Science DOI: 10.1126/science.aah4427) |
| |
|
| Ward等人利用定向突變策略開發了數以千計的蛋白質變體,并選擇了最活躍的一種能夠催化細胞中的復分解反應。Roelfes等人開發了人工銅-聯吡啶催化劑,通過在體內將金屬結合、非天然氨基酸結合到蛋白質支架中,或通過創建與銅結合的人工活性位點,實現選擇性Friedel-Crafts烷基化。(Nature 537, 661–665 (2013); Chem. Sci. 6, 770–776 (2015)) |
| 盡管在將單個催化實體引入蛋白質支架方面進行了大量的努力,但在引入多個活性位點以及分析這類活性位點的催化優勢這方面的研究十分罕見。 |
| |
|
| 構建兩個血紅素、Fe-S或銅位點的蛋白質支架;催化金屬引入脂肪酶。雖然賦予級聯反應的能力,但催化過程分兩步進行,改變了反應條件,排除了協同效應。(Nat. Chem. Biol. 9, 826–833 (2013). Angew. Chem. Int. Ed. 54, 511–515 (2015)) |
| 酶工程不僅可以從頭開始創造活性位點,以接近擴散極限的速率催化已知的生物反應,而且還可以生成進行新的自然反應的非生物位點。然而,將多個活性位點工程化為單個蛋白支架的催化優勢尚未確定。 |
| |
|
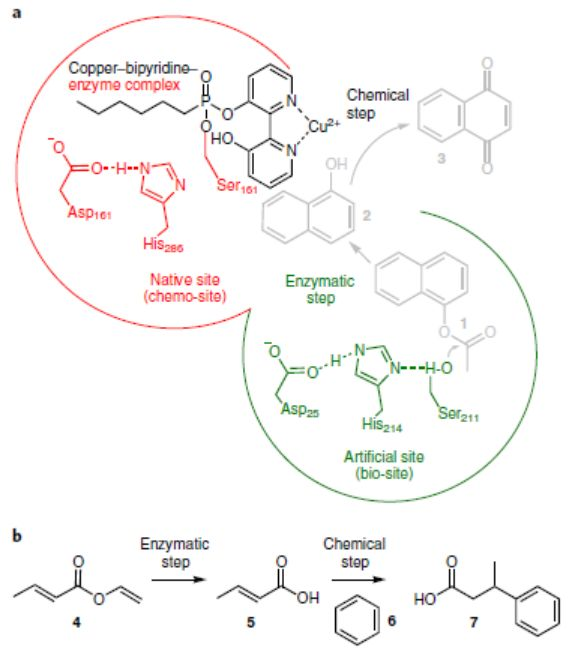
| 近日,西班牙高等科學研究理事會Manuel Ferrer教授、巴塞羅那超級計算中心Víctor Guallar教授、西班牙物理化學研究所Julia Sanz-Aparicio教授合作利用酶工程技術構建了一種具有兩個生物/非生物起源的活性位點的人工金屬酶,以改進天然和非天然催化作用。(圖1)(1)該方法大大提高了含有兩個活性位點酶的酶效率、底物范圍、立體選擇性和最佳溫度窗口等催化性能。(2)將其中一個活性位點轉化為金屬絡合物的化學催化位點,進行氧化和Friedel–Crafts烷基化反應,促進單一蛋白質中的協同化學和生物催化。(3)利用這種多功能金屬酶,可將乙酸萘酯轉化為1,4-萘醌(轉化率約100%),將巴豆酸乙烯酯和苯轉化為3-苯丁酸(轉化率≥83%;e.e.>99.9%)。(4)該方法在制備具有改進的催化性能的生物催化劑方面,以及在制備生物和非生物催化實體協同作用下進行級聯反應的金屬酶方面,均具有巨大的潛力和通用性。 |
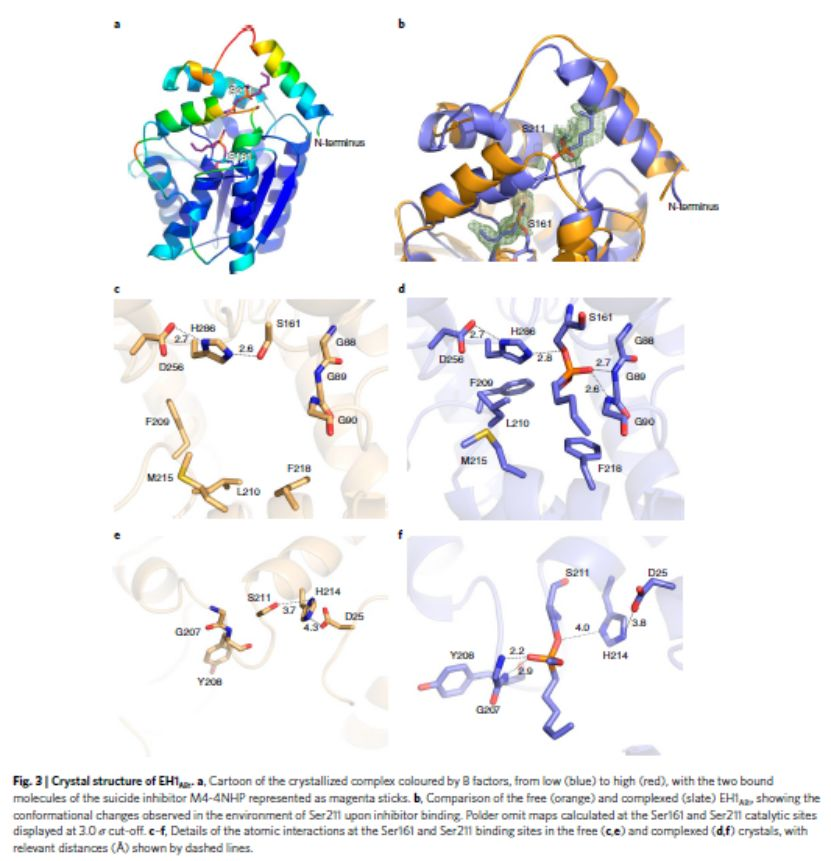
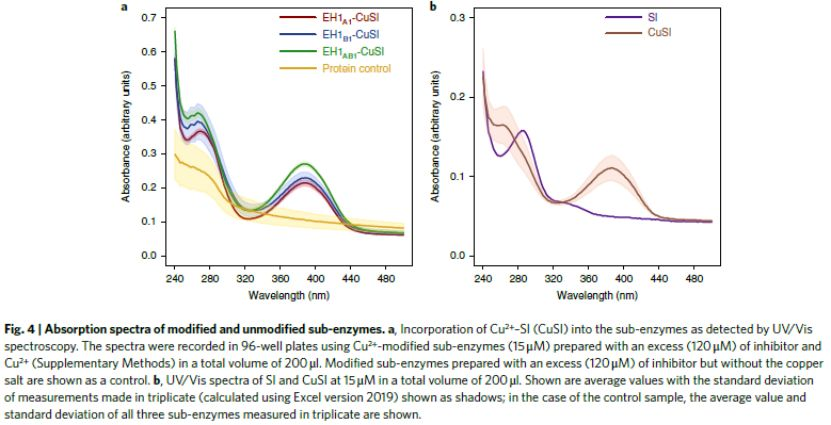
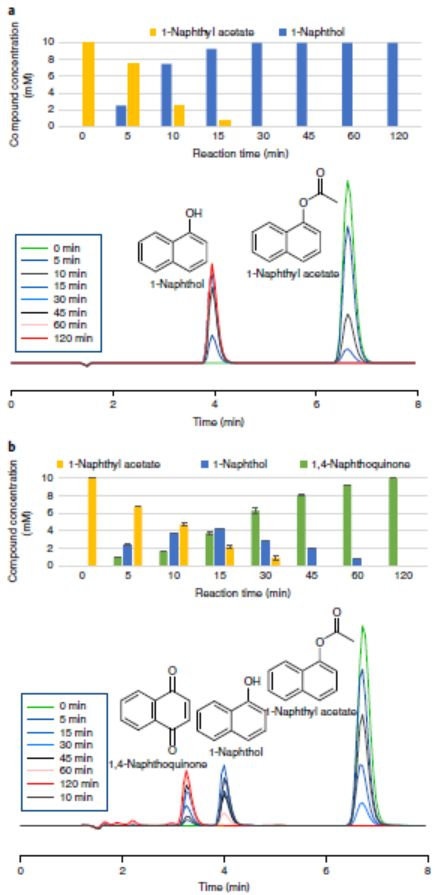
酶工程可從頭創造活性位點,而且還可以生成進行新的自然反應的非生物位點,因此,利用酶工程制備可媲美天然酶催化性能的人工酶具有重要意義。其中人工金屬酶(Arms)由于其優異的催化性能備受關注。目前的研究集中于構建單一活性位點的Arms且催化性能不如天然酶。所以,研究人員提出構建多活性位點的Arms,這類設計雖然賦予Arms級聯反應的能力,但催化過程是分兩步進行,改變了反應條件,排除了協同效應。首先,利用酶工程技術構建具有兩個活性位點的Arms,然后,通過理化性質表征Arms催化優勢,最后,利用兩個化學反應驗證其協同催化優勢。首先,構建具有兩個活性位點的Arms(如圖1所示)。1)選擇含有生物活性位點(生物位點1)的目標天然酶(如EH1A1)。2)通過應用蛋白質能量景觀探索(PELE)軟件確定另一個結合袋,利用酶工程技術引入一個人工生物活性位點(如EH1B1),結合袋可以被進一步改造以獲得最佳配置(生物位點2),得到EH1AB1。3)通過對含有金屬-有機絡合物的自殺性膦酸鹽抑制劑親和力的差異,一個生物位點被轉化為銅基化學催化位點(化學位點),而另一個位點保留其生物活性(生物位點),得到最終的金屬酶(EH1AB1C-B)。通過將第二活性位點EH1B1引入天然酶EH1A1中,利用反應常數(kcat)、催化效率(kcat/Km)、立體選擇性(e.e.)、酶的最佳活性溫度等指標來判斷EH1B1的引入是否增強EH1A1的催化性能。EH1B1與EH1A1結合增加了kcat(平均約3.4倍;最大約74倍)和kcat/Km(平均約94倍;最大約5000倍)酯水解值(圖2a-2b),立體選擇性增加約1100倍(e.e.>99.9%)(圖2c),并使酶保持其最佳活性80%以上的溫度增加約20℃(圖2d)。結果表明,在特異性、親和力、轉化率和局部穩定性方面,EH1B1的引入顯著增強天然酶的原始生物催化特性。為了證明EH1AB1的兩個活性位點都能夠與底物結合,并且可以在兩個位點發生轉化,作者EH1AB1進行了結構分析。從EH1AB1獲得了分辨率為2.1?的晶體。這些晶體與抑制劑4-硝基苯基己基膦酸甲酯(M4-4NHP)共結晶得到相應的衍生物絡合物,抑制劑的兩個分子與催化絲氨酸殘基Ser161(原始親核劑)和Ser211(人工親核劑)結合(圖3a)。所求解的三維結構在含有兩個N末端螺旋區域顯示出高度的靈活性,這兩個N末端螺旋基序允許進入活性位點口袋。如圖3b所示,在抑制劑結合后,在二級Ser211催化位點(人工重塑位點)觀察到構象變化,這將扭曲浸透晶體內的排列。結合到兩個位點的抑制劑的原子相互作用可以被描繪并且顯示在圖3c-f中。(自由晶體(c,e)和復合晶體(d,f)中Ser161和Ser211結合位點的原子相互作用的細節)3)EH1A1、EH1B1和EH1AB1均能與Cu-SI緊密結合。作者發現天然酶(Ser161)和人工酶(Ser211)均能與M4-4NHP抑制劑結合,因為不到10分鐘,用過量SI處理EH1A1、EH1B1和EH1AB1可導致>99%的酶失活。隨后作者合成過渡金屬螯合M4-4NHP的自殺抑制劑(Cu-SI),賦予緩蝕劑單元過渡金屬催化功能,成為亞酶的兩個活性位點之一,而另一個保持完整。為了驗證這種生物結合,將SI修飾的亞酶與過量的Cu(NO3)2孵育24小時,然后透析去除未偶聯的抑制劑和銅,并通過UV-Vis進行分析。在UV-Vis光譜中觀察到,EH1A1-SI、EH1B1-SI、EH1AB1-SI在386nm處存在λmax(圖4a)這是Cu2+聯吡啶絡合物的特征(圖4b);在沒有Cu2+的情況下,沒有檢測到蛋白質的這種信號(圖4a)。除了UV-Vis光譜分析,還對Cu2+有機配合物的氧化還原性質和ESI-MS對其進行了分析。結果表明,天然酶(Ser161)和人工酶(Ser211)均能與M4-4NHP抑制劑(SI)緊密結合。1)EH1AB1C-B及其在兩個模型反應中的應用。EH1AB1C-B的催化活性是通過酯的酶解反應和雙吡啶銅氧化反應(圖a)或Friedel-Crafts烷基化反應(圖b)兩種級聯反應來評估。由于酯1的高轉化率和雙吡啶銅的電子轉移能力,作者選擇了這兩個反應作為第一個級聯反應。在圖a中,酯1的酶解產生醇2,而醇2可能被雙吡啶銅催化劑氧化為醌3。雙吡啶銅催化劑已被證明可以進行Friedel-Crafts烷基化反應,同時,酯4在人工位點容易轉化,由此產生的水解產物可以與苯偶聯,所以選擇了這兩個反應作為第二個級聯反應。在圖b中,酶水解的酯4將生成烯基脂肪酸5、酸5與苯6產生酸7。2)驗證EH1AB1C-B催化乙酸萘酯生成1,4-萘醌第一個反應條件為pH 8.0和25℃,酯110mM,使用不含Cu2+的修飾蛋白,未形成醌3,但觀察到醇2的形成(100%轉化率)(圖a)。結果符合預期,因為生物催化位點是唯一的活性位點。然而,在化學-生物催化劑(EH1AB1C-B)的存在下,經過2小時的反應,醌3的轉化率達到100%(圖b)。3)驗證EH1AB1C-B催化乙烯基丁酸與苯一鍋法合成3-苯基丁酸第二個級聯反應在4℃下反應3天。使用不含Cu2+的修飾蛋白,僅生成烯基脂肪酸5,未消耗苯6(圖a),而在化學-生物催化劑(EH1AB1C-B)的存在下,3天后轉化為產物7的比例達到83%(圖b)。