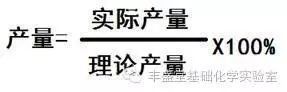
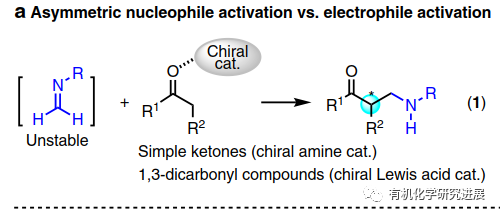
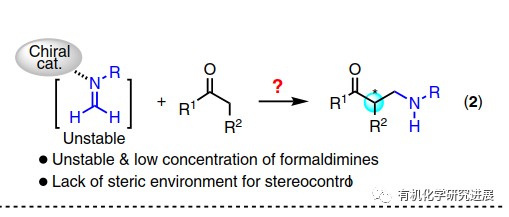
作為曼尼希反應的一個重要的分支,羰基化合物的α-胺甲基化是引入胺甲基到簡單有機分子的一種強大的方法,生成的β-胺基羰基化合物可以用于合成許多天然產物和生物活性分子。一般來說甲醛衍生的亞胺或者亞胺鹽都不太穩定,通常是由甲醛與芳香胺反應,或者由α-氨基甲基醚,N,O-乙縮醛,1,3,5-三芳基-1,3,5-三嗪原位生成。在此背景下,通過胺或手性Lewis酸催化劑不對稱活化親核性的羰基化合物已經被用于實現該類轉化的不對稱轉化。雖然活化親核試劑的策略可行并且有較好的對映選擇性,但是該親核試劑通常限定于本身較為活潑的底物,如:未修飾的酮和1,3-二羰基化合物。另一方面,雖然手性Br?nsted酸活化穩定的亞胺的策略已經被廣泛應用到不對稱曼尼希反應,但是這種親電不對稱活化的策略目前尚未被應用到原位生成的甲醛亞胺的胺甲基化反應中,可能歸因于甲醛亞胺的不穩定的性質以及碳原子上缺少有效的空間位阻環境無法實現更好的立體選擇性控制。
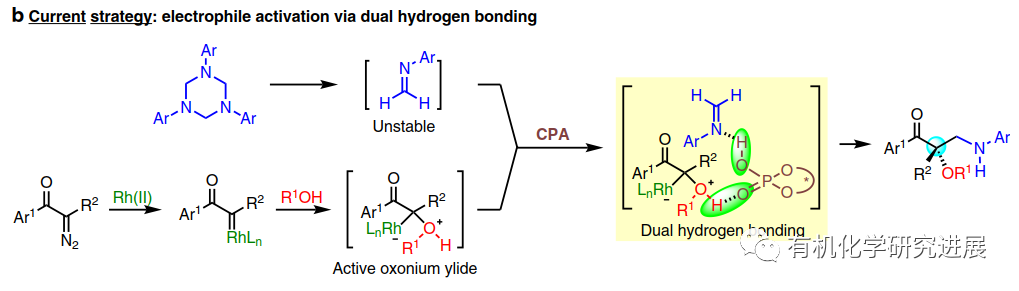
近些年,胡文浩課題組已經證實基于活性鎓葉立德親電捕獲的金屬卡賓參與多組分策略能很好的實現以前反應低效甚至不可能的反應。而這其中,曼尼希類型的芳亞胺捕獲氧鎓的反應可以快速合成β-氨基醇。在銠/手性磷酸(CPA)的協同催化下,CPA在氧鎓和亞胺底物上的雙氫鍵活化很好的保證了該反應的立體控制(10.1002/tcr.201600124)。
受此啟發,該課題組設計了一種α-重氮酮、醇、1,3,5-三芳基-1,3,5-三嗪參與的三組分反應,期望通過類似的雙氫鍵活化模式,從而能實現甲醛亞胺的親電不對稱活化發生不對稱胺甲基化反應,合成一系列手性β-胺-α-羥基酮,這類化合物廣泛存在合成以及藥物領域(Nat. Commun. 2020. 10.1038/s41467-020-15345-2)。
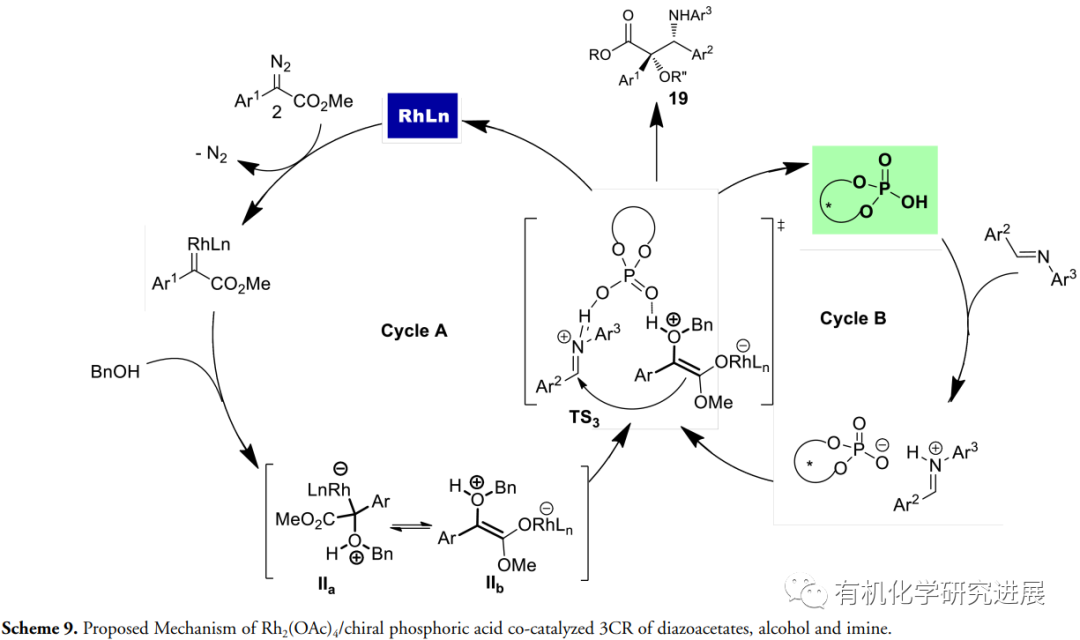
設計這種三組分反應有三個挑戰:(1)1,3,5-三嗪原位生成的醛亞胺在反應體系中的濃度很低,因此親核試劑捕獲氧鎓變得很低效,更傾向于得到非目標的O-H插入產物。(2)1,3,5-三芳基-1,3,5-三嗪在金屬催化下可能與重氮進行[4+1]環加成反應,降低目標的三組分反應產物的產率;(3)還沒有實驗證實是否CPA和兩個活性中間體能有效的形成雙氫鍵,這也是由于它本身不穩定的性質、反應過程中的濃度低、原位生成醛亞胺的碳原子上缺少取代基。
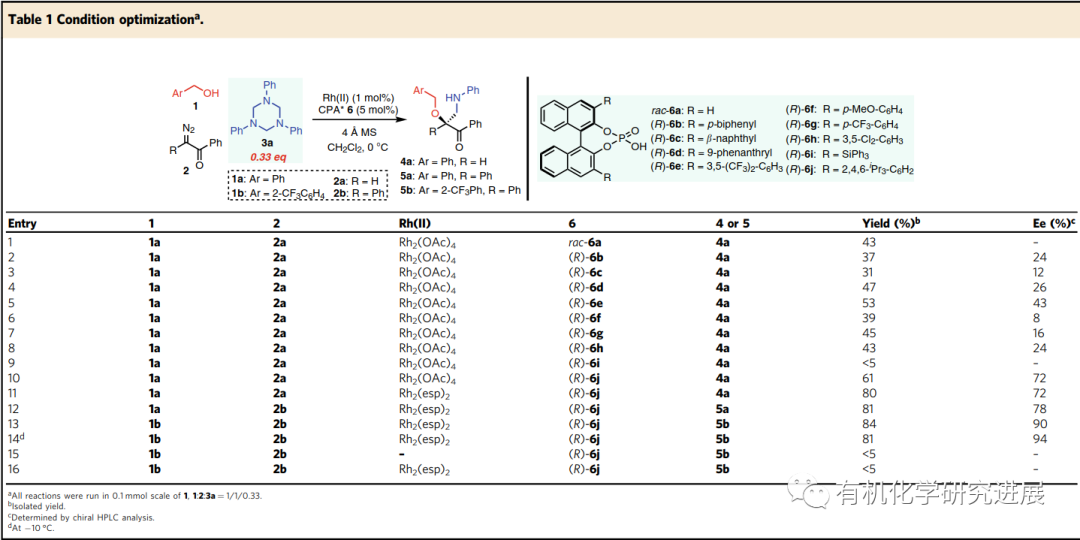
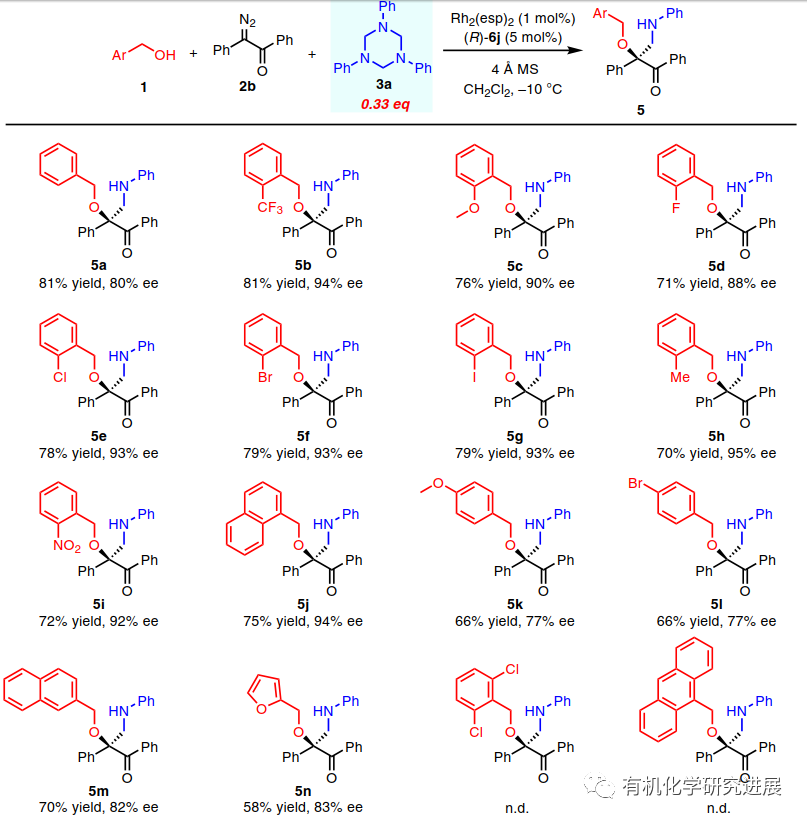
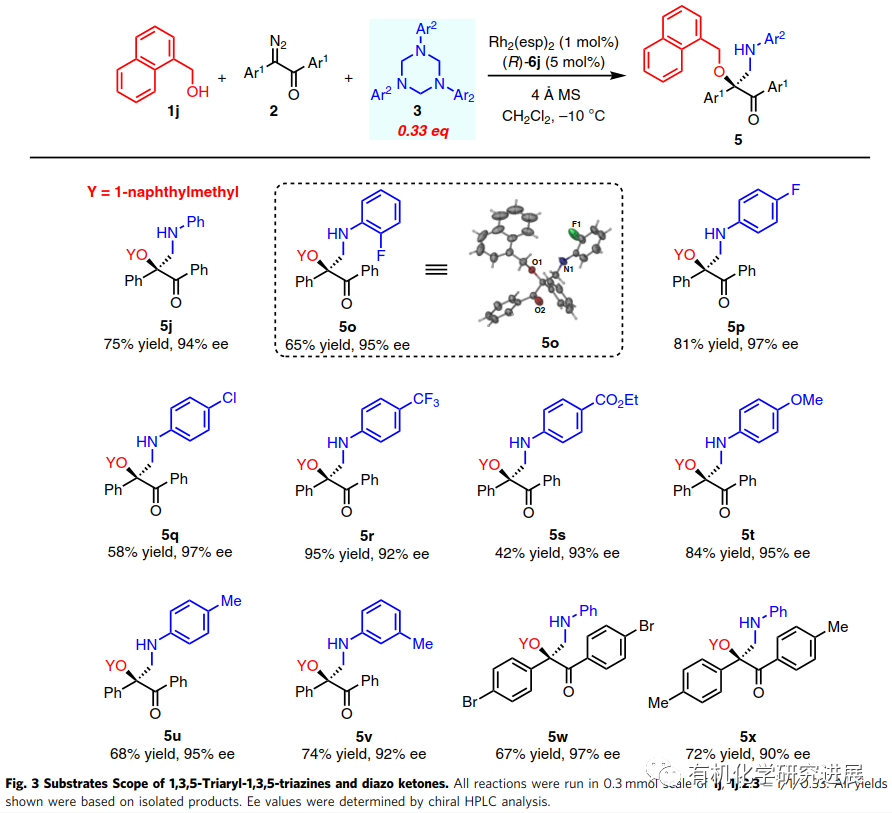
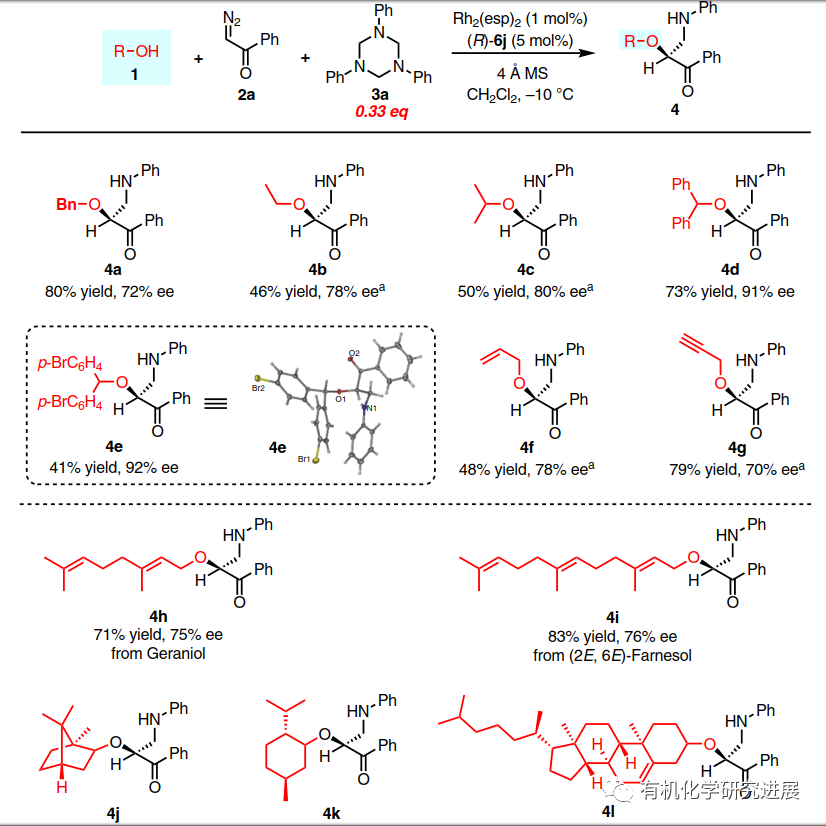
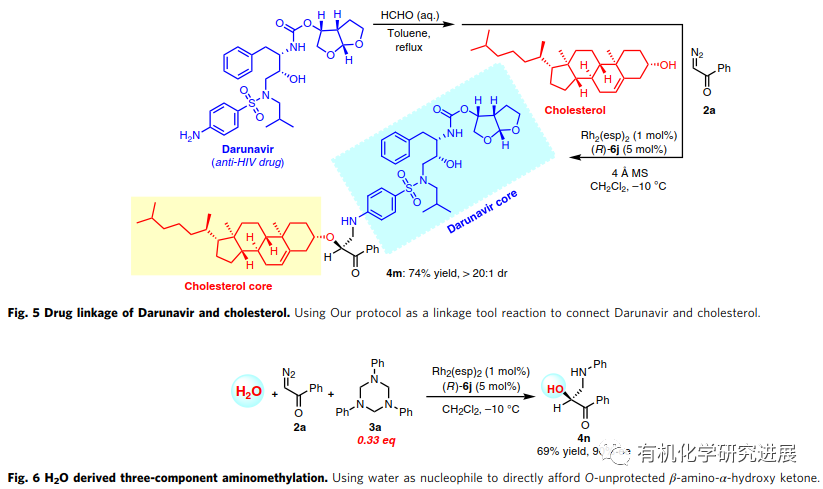
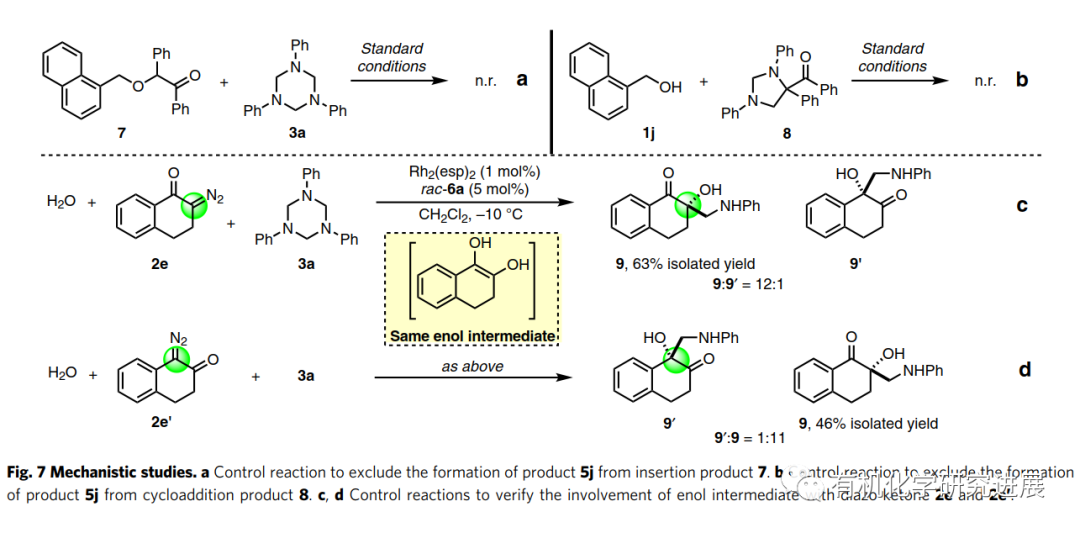
首先,該作者選擇以1,3,5-三芳基-1,3,5-三嗪3a作為甲醛亞胺的前體,在銠(II)/磷酸共同催化下優化不同的重氮酮和芐醇1a的三組分反應。控制實驗表明Rh(II)和CPA都是必不可少的。當缺少CPA時,檢測不到三組分反應的產物,相反2a和3a的[4+1]環加成反應成為了主反應。 在得到最優反應條件之后,作者首先對芐醇的底物范圍進行了探究。結果表明芐醇底物的位阻比電子密度對反應的對映選擇性有更大的影響。進一步增加醇化合物的位阻導致無法得到目標產物,這表明轉化的發生和位阻之間存在一個嚴格的平衡。 三嗪的底物范圍較廣,電子效應和位阻效應在對映選擇性方面不明顯。 由于手性β-氨基-α-羥基酮有很好的合成潛力,作者又繼續擴大醇的底物范圍。烷基醇、烯丙醇和炔丙醇也能順利的反應,其中烯丙基和炔丙基有利于進一步衍生化β-氨基酮。該方法還可以對各種天然產物的醇類化合物進行后期修飾。 商品化藥物的修飾和兩個藥物結構的連結對于發現新的藥物分子有很大的作用。利用該策略將抗HIV藥物Darunavi和膽固醇連結,能高產率和非對映選擇性的生成新化合物4m。更驚奇的是,水也可以作為親核試劑參與反應生成手性β-氨基-α-羥基酮。 為了探究該反應的機理,作者進行了兩個控制反應。首先,在標準條件下,將單獨制備的O-H插入產物7 與3a進行反應,該反應無法進行。這個結果表明該轉化不是經歷O-H插入/曼尼希反應的分步路徑。另一方面,作者將只使用銠(II)催化劑時生成的[4+1]環加成產物8,在標準條件下與1j反應,該反應同樣無法進行。因此也排除了8作為轉化反應中間體的可能。 此前已有報道提出烯醇中間體是α-重氮酮和醇的O-H插入和重排反應的關鍵中間體,相關的DFT計算也支持了這一可能。為了證明烯醇是否能參與本轉化,作者分別使用2-重氮-1-四氫萘酮和1-重氮-2-四氫萘酮進行了平行反應,都得到相同的主產物,這表明了是烯醇中間體而不是氧鎓參與了該轉化。 總結,在銠(II)/手性磷酸催化下,胡課題報道了一種由α-重氮酮、1,3,5-三嗪、醇三組分參與的不對稱催化反應。通過甲醛亞胺的親電不對稱活化實現不對稱胺甲基化反應,合成一系列手性β-胺-α-羥基酮化合物。各類的醇包括簡單的脂肪醇,復雜的天然產物醇和水都可以進行反應,反應的效率高且對映選擇性好。