在眾多ent-貝殼杉烯二萜類化合物中,(-)-oridonin(2)是臨床和臨床前研究中應用最廣泛的分子之一。事實上,它是常用的非處方抗炎中藥冬凌草的主要活性成分。二萜化合物2在體外和體內表現出對多種癌細胞的潛在活性,這使得研究人員對其進行大量的藥物化學研究,并微調了一系列oridonin衍生物的特性以用于藥物開發。盡管化學家們已經完成了oridonin的半合成,并且在化學合成高氧化的ent-貝殼杉烯二萜類化合物方面取得了重大進展,但(-)-oridonin(2)的全合成仍充滿挑戰,因此發展新型、有效的合成策略仍是必要的。近日,北京大學羅佗平教授課題組利用一個關鍵的中斷Nazarov反應實現了(-)-oridonin(2)的對映選擇性全合成。相關研究成果發表在J. Am. Chem. Soc.上(DOI: 10.1021/jacs.9b12034)。
逆合成分析如Figure 1所示,后期中間體3可以由環氧化物4經過1,2-遷移和脫保護來制備。另一個1,2-遷移(偶姻重排)可以將4追溯到5。該縮環策略展現關鍵的合成中間體6。作者設想了一個中斷的Nazarov環化反應,該反應利用側鏈烯丙基硅烷捕獲羥基烯丙基陽離子7a以有效合成6。該關鍵反應的底物酮8a可以由兩個單環合成子9和10制備。
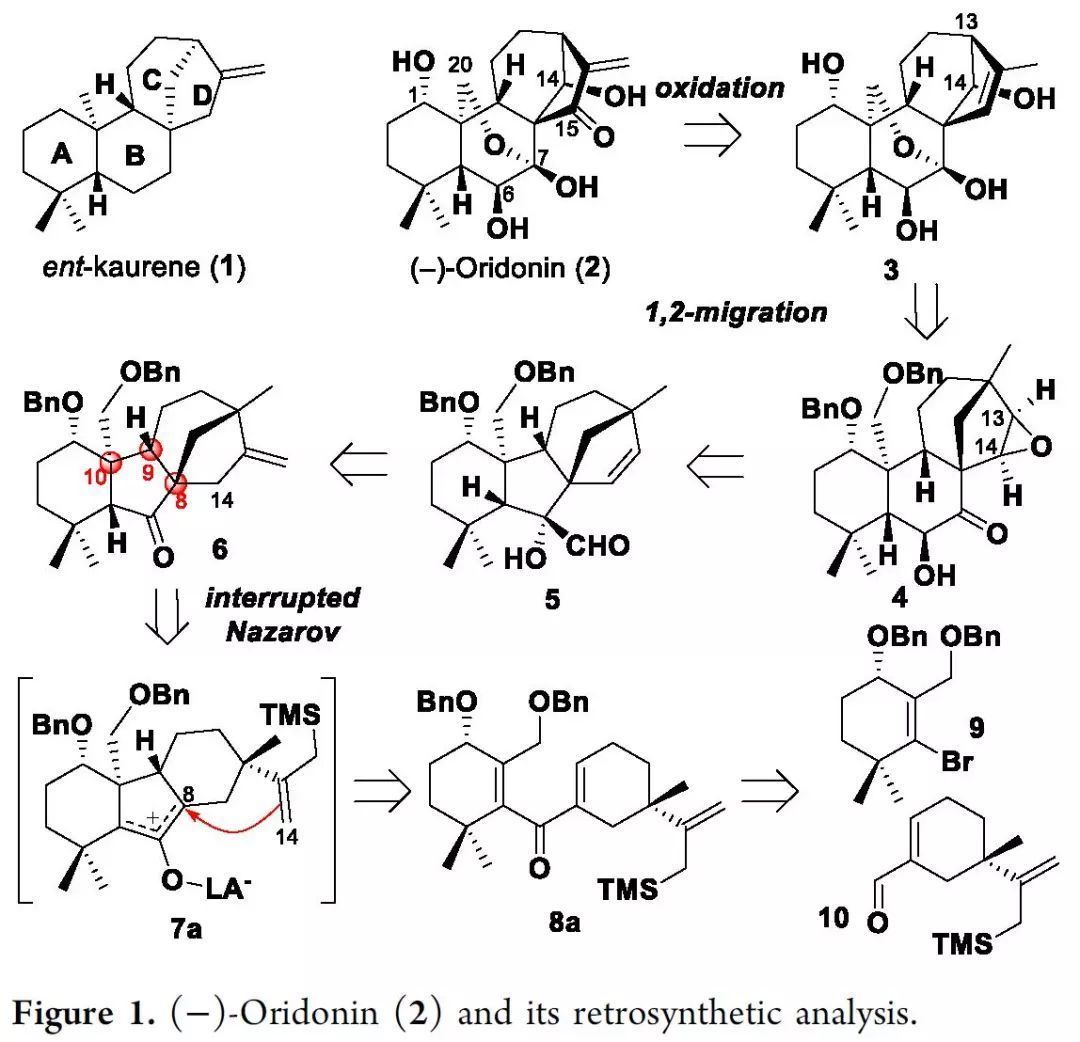
(來源:J. Am. Chem. Soc.)
作者首先制備了非對映異構體混合物為1:1的酮8(Table 1),以評估(C1和C16)立體化學對Nazarov/Hosomi-Sakurai串聯反應的影響。通過對路易斯酸的篩選,作者發現僅EtAlCl2有利于反應的轉化。所得的產物(11-14)表明此反應的兩個重要特征:(1)可以通過C1和C20 芐基保護基來調節產物的旋光選擇性;(2)反應是以立體選擇性的方式形成了中斷的Nazarov反應的產物。隨后經溶劑等條件的進一步優化,在克級規模下產物11和14的收率分別達到>30%和>20%(entries 6和7)。
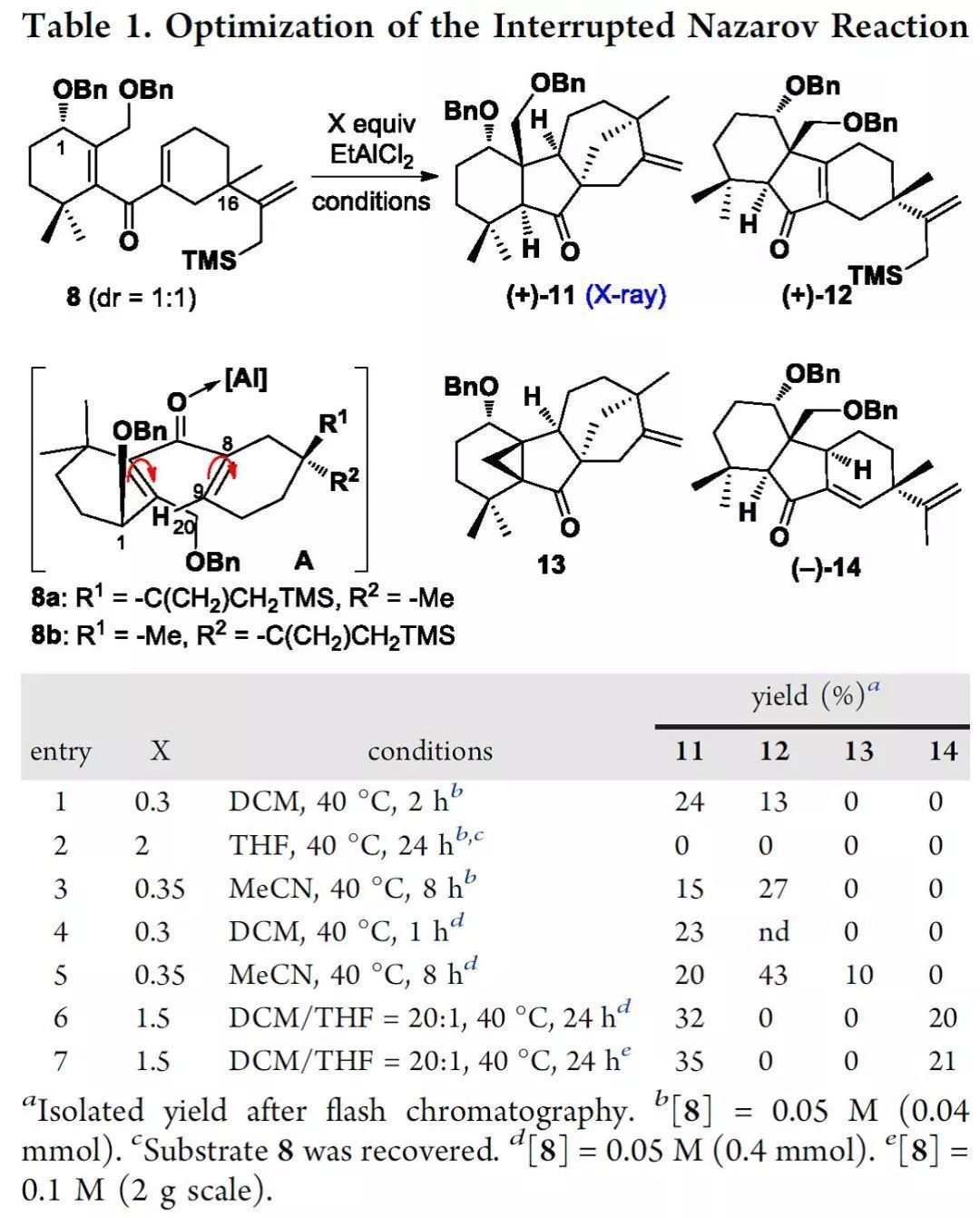
(來源:J. Am. Chem. Soc.)
基于對Nazarov/Hosomi-Sakurai串聯反應的研究結果,作者調整了合成路線以制備(+)-9(Scheme 1)。酮15經Vilsmeier反應得到β-溴醛16,其隨后經Pinnick氧化和甲基化轉化為酯17。通過篩選各種烯丙基氧化條件,作者發現CrO3可以在20 g規模下以45%的收率生成烯酮18。化合物18在過量的BMS(硼烷二甲硫醚絡合物)和CBS(2-甲基-CBS-惡唑硼烷)作用下直接以優異的對映選擇性還原得到手性二醇(+)-19,其經芐基保護得到(+)-9。
接著,作者從(+)-20開始合成右側片段10。酮20經一氯化碘處理轉化為碘化乙烯21,然后其與三甲基甲硅烷基甲基鋅試劑進行Pd催化的交叉偶聯反應,以兩步55%的收率得到烯丙基硅烷22。22上羰基空間位阻較小的α-亞甲基經區域選擇性脫質子反應生成了三氟甲磺酸烯醇酯,其經鈀催化的羰基化反應以兩步63%的收率得到酯23。23轉化為Weinreb酰胺24,然后經DIBAL-H還原,得到對映體富集的醛(+)-10。
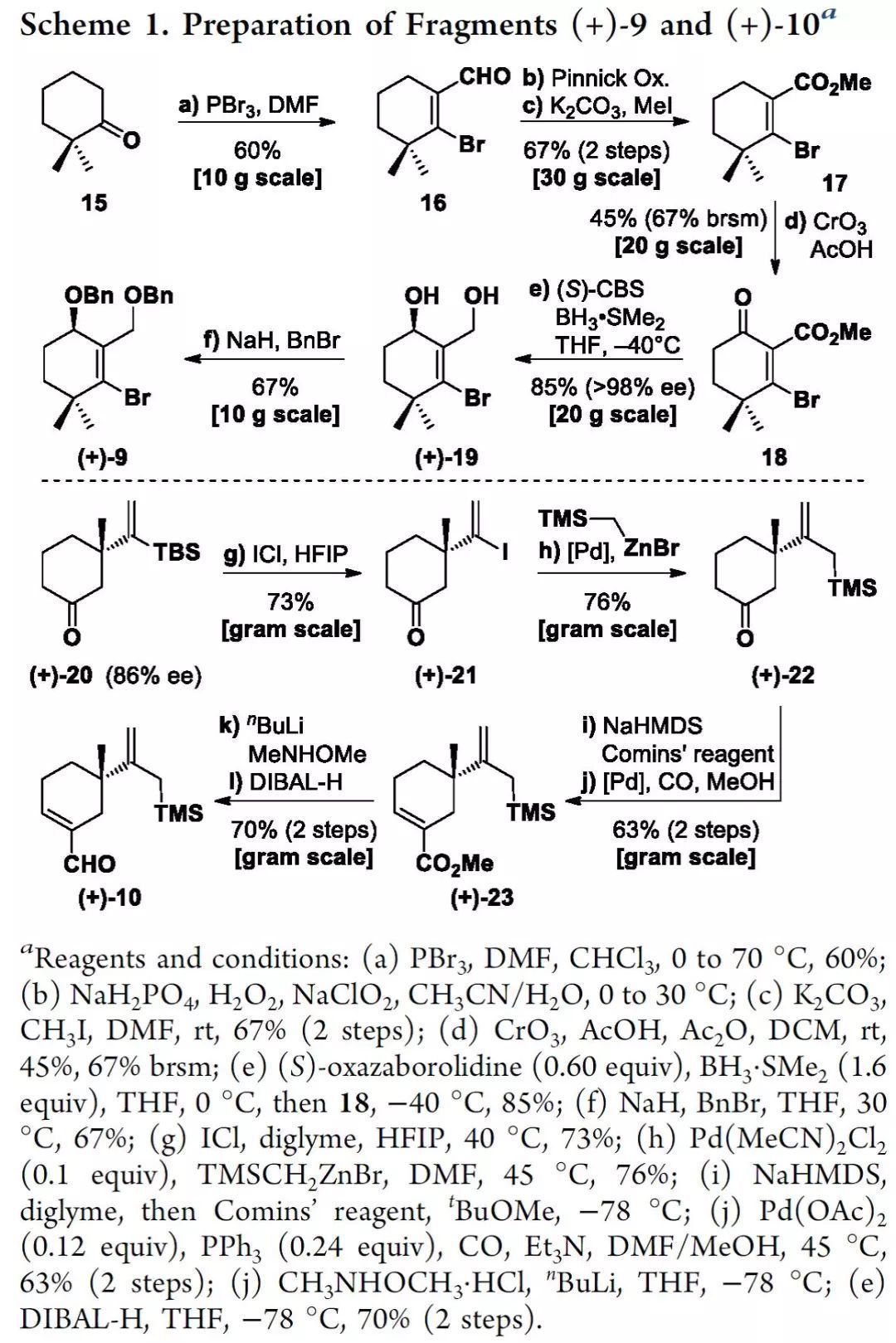
(來源:J. Am. Chem. Soc.)
得到上面兩個片段后,作者繼續進行(-)-oridonin(2)的全合成(Scheme 2)。(+)-9和tBuLi經Li-Br交換制備的鋰試劑與(+)-10加成得到一對非對映體仲醇,然后經PDC氧化形成(+)-8b。在Nazarov/Hosomi-Sakurai串聯反應的優化條件下,(+)-8b轉化為(-)-11。所得的粗產物直接進行單線態氧烯反應,然后用乙酸酐處理,以兩步43%的收率得到醛(+)-25(J. Org. Chem. 1983, 48, 4135?4137.)。隨后醛25經銠催化的去甲酰基化反應,克級規模下以67%的收率得到酮26。在篩選各種親核試劑后,作者利用乙烯基鋰實現了1,2-加成反應,生成的醇上C13-C14烯烴經間氯過苯甲酸選擇性環氧化,得到單一非對映異構體27。碳碳雙鍵上首先引入親電溴,接著經類頻哪醇重排反應構建了B環,進而得到28,其結構由X射線衍射確定。為避免在路易斯酸條件下C20-OBn基引起的環氧化物開環以及與不穩定溴化物相關的副反應,作者首先對28進行了一系列的氧化轉化。兩個芐基保護基被轉化為苯甲酰基,然后經DBU處理得到烯酮29,其經雙羥基化反應以兩步70%的收率獲得30。經過對路易斯酸篩選,作者發現EtAlCl2可有效促進關鍵的環重排反應,從而構建了C/D環系。LiAlH4還原酮31的C6羰基、同時去除兩個苯甲酰基保護基得到己醇32,其粗產物經選擇性裂解空間位阻較小的1,2-二醇以兩步69%的收率得到半縮醛33。
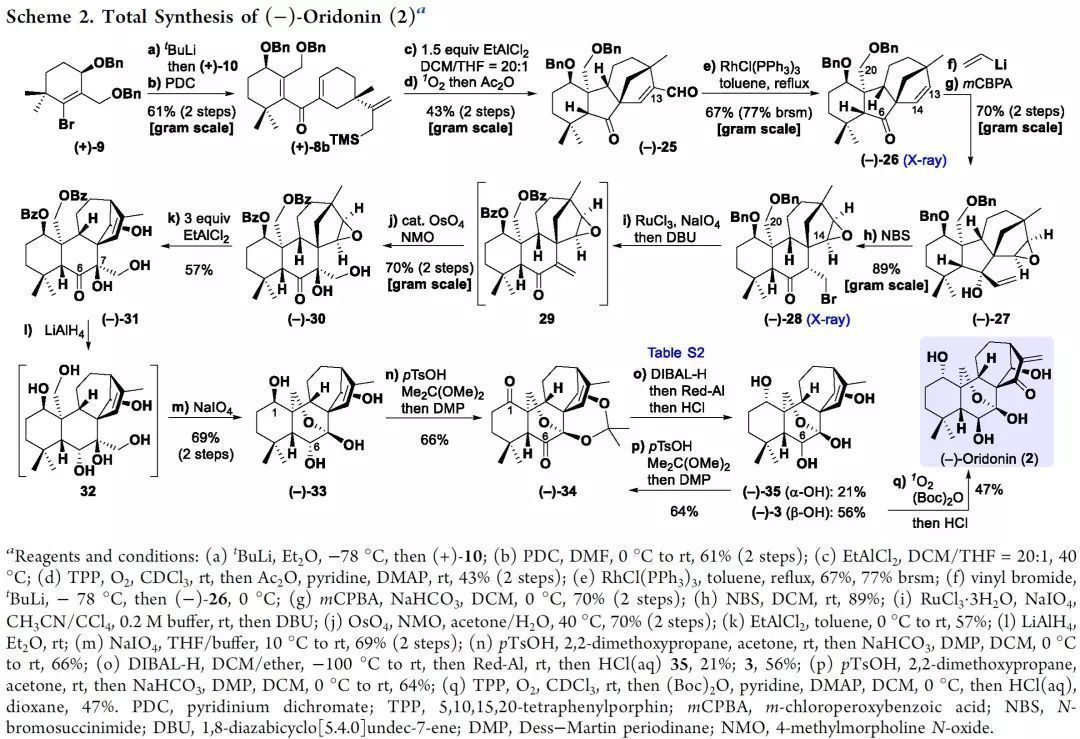
(來源:J. Am. Chem. Soc.)
中間體33經丙酮類似物保護,隨后進行Dess-Martin氧化,一鍋法以66%的收率形成酮34。通過仔細優化還原/脫保護順序和反應條件,作者首先用DIBAL-H在-100 °C下于DCM/乙醚(3:1)中區域和非對映選擇性還原C1酮,然后用Red-Al還原C6羰基,從而在原位脫保護后分別以21%和56%的收率得到非對映異構體(-)-35和(-)-3。3進行單線態氧烯反應,然后經TCCA處理,可以形成烯酮片段,但僅能以較低的收率得到(?)-oridonin(2),最終,作者發現(Boc)2O能有效地將衍生自(-)-3的烯丙基氫過氧化物轉化為烯酮,同時保護C14羥基。因此,隨著HCl的加入,該一鍋法反應能以47%的收率得到天然產物2。
總之,作者首次實現了(-)-oridonin(2)的全合成。此項工作進一步證明了中斷的Nazarov環化反應在全合成中極具應用價值。該策略的另一個亮點是通過兩次1,2-遷移轉化進行的關鍵骨架重排。此外,經克級規模制備的兩個合成子(+)-9和(+)-10也可以用于合成其他萜類化合物。








