

▲論文DOI: 10.1016/j.apcatb.2020.118855,
第一作者:李永利; 共同通訊作者:北京工業大學王金淑教授和澳大利亞昆士蘭大學王連洲教授。
研究背景
光催化分解水是應對未來可持續能源存儲和轉化的一個重要途徑。太陽能燃料轉換(STF)效率取決于光的吸收/激發效率、電荷分離/遷移效率以及表面氧化還原反應速率。作為一種廉價的有機聚合物半導體,類石墨相氮化碳(C3N4)具有比較合適的能帶結構,理論上能夠利用可見光激發分解水產生氫氣和氧氣。然而,大多數情況下,由于C3N4層間π-π堆積和層內較低的電子傳輸特性致使材料的性能受限于以上的一個或多個步驟。通過物理結構和電子結構的調控,增加催化位點數量,加速電子遷移/擴散動力學,是提高光催化性能的有效手段。
A. C3N4的物理結構調控
對于包括C3N4在內的二維半導體來說,對體相材料進行剝層是增加催化位點數量、縮短載流子遷移距離的最有效的方法之一。目前,無論采取自上而下(top-down)還是自下而上(bottom-up)的路線都可以將體相C3N4剝層至單原子層或幾個原子層厚度。其中,自上而下的路線因具有靈活多變的處理工藝和不同的插層/鈍化劑可供選擇,因此受到大多數研究者的青睞。另一方面,剝層也帶來了一些不利的后果,致使剝層后的光催化性能仍不如人意:①量子限域效應變得更加顯著,帶隙(Eg)變寬不利于對太陽光的利用;②剝層導致C3N4表面/邊緣基團發生不可控的改變,易成為載流子的復合中心。
B. C3N4的電子結構調控
對共軛結構進行原子置換或基團接枝,尤其是C或N原子自摻雜,可以有效地調節C3N4的電子結構以提高共軛結構內電子遷移速率、窄化帶隙,近年來備受矚目。其中包括由C取代引起的π電子注入(Angew. Chem. 2019, 131, 2007;Adv. Mater. 2018, 30, 1705060)、C接枝構建的層間復合與層內復合(Science, 2015, 347, 970;J. Am. Chem. Soc. 2017, 139, 3021)等。
C. 目前研究的局限性
各類已經報道的對于C3N4改性的研究,大都著眼于單一的物理結構或電子結構調控,通過增加活性位點數量或加速載流子遷移速率,以期實現更高的催化活性。兩種調控方式均有各自突出的優點,但也存在明顯的不足。如何協調光的利用效率和催化活性位點數量,在提高載流子遷移速率的同時縮短遷移距離,仍然是一個難以兼顧的硬幣的兩面。
催化劑的設計與制備
基于前期研究和上述分析,北京工大學王金淑課題組和澳大利亞昆士蘭大學王連洲課題組合作,提出一種連續后氧化還原(Post-redox)策略對C3N4進行原位剝離和C取代,同時調控物理結構和電子結構,以期擁有二者的協同優勢。所采用的Post-redox策略包括兩個步驟,如Fig. 1所示。首先在水熱條件下利用堿溶液輔助水解和插層作用,對層間剝離并打開橋連的N-3C位,使C6N7共軛環端部羥基化,此過程同時也刻蝕去除了不完全聚合的部分結構以使空腔擴大;其次,引入羥基功能化的C團簇,通過氫鍵與氧化的C3N4結合;最后通過熱處理,出去多余的O、C,使結構穩定化,得到橋連位C取代的介孔C3N4超薄納米片:CRed-AT-C3N4。
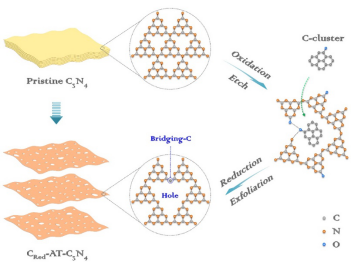
▲Fig. 1. Schematic representation for the formation of CRed-AT-C3N4.
催化劑的結構表征
所制備的CRed-AT-C3N4形貌結構信息如下。呈現出富含雙尺寸介孔的超薄納米片,片層厚度約為1 nm,相當于3-4層原子厚度。并且經過一系列處理,C、N元素分布均勻,未出現富C區(Fig. 2)。
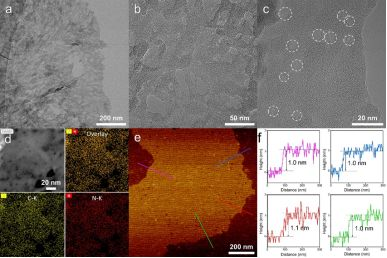
▲Fig. 2. (a-c) TEM image of the CRed-AT-C3N4 and (d) elemental distribution mapping. (e, f) AFM image of the CRed-AT-C3N4 and height profiles along with the corresponding lines in (e).
C 對橋連N的取代由如下表征手段加以證實。首先,材料的孔結構和外觀顏色雖然發生了顯著的改變(由淡黃色變為棕色),但C3N4的基本化學結構并未受到破壞,而FTIR譜中出現了C-H和C=C振動峰,說明C進入C3N4分子內形成摻雜態(Fig. 3)。同時,電子結構也發生了一些精細的變化。X射線近邊吸收譜(XANES)顯示,在共軛結構內形成了新的sp2-C。而N K-edge譜中橋連N-3C峰強度的減弱正是由于C的取代所引起。13C核磁共振譜(NMR)和XPS中端基-NHx的減弱也側面證實了橋連N-3C的取代。取代的結果使得C6N7共軛單元的π電子密度增加,這有利于加速電子在層內的遷移(Fig. 4)。
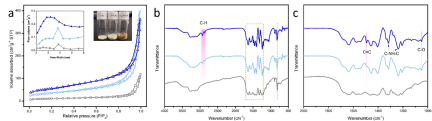
▲Fig. 3. (a) N2 adsorption-desorption isotherms of pristine C3N4 (gray), AT-C3N4 (azure) and CRed-AT-C3N4 (blue). Inset is the pore size distribution. (b, c) FTIR spectra of pristine C3N4, AT-C3N4 and CRed-AT-C3N4.
作者進一步運用密度泛函理論進行了計算。計算結果表明,橋連N-3C的取代使共軛環上的電子更加離域化,說明在CRed-AT-C3N4中HOMO電子更容易受激發躍遷至LUMO,載流子的空間分離也更加顯著。光學/光電分析結果與計算結果同樣表現出很好地契合。帶隙變窄,載流子壽命增加,電子-空穴復合受到有效地抑制,改善了載流子的遷移效率,這些結果表明作者提出的Post-redox策略很好地協調了原子層厚度與電子結構的優點。
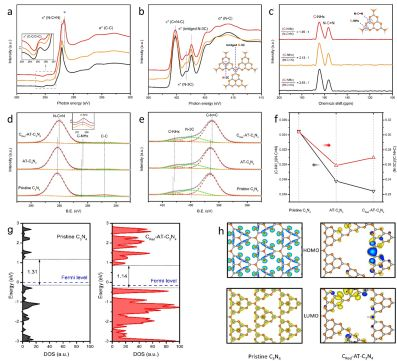
▲Fig. 4. (a, b) XANES of C K-edge and N K-edge of pristine C3N4 (black), AT-C3N4 (orange) and CRed-AT-C3N4 (red). (c) Solid-state 13C MAS NMR spectra. (d, e) XPS C 1s and N 1s, (f) surface structural features according to the XPS peak area ratios. (g, h) Calculated density of states and spatial distributions of HOMO (blue) and LUMO (yellow).
光學測試結果說明(Fig. 5),CRed-AT-C3N4具有變窄的帶隙和提高的載流子傳輸速率,光生電子的壽命得到延長,即,載流子更為有效地分離,很好地支持了計算結果。
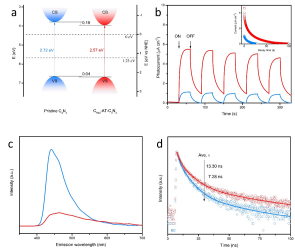
▲Fig. 5. (a) Band structure diagrams of pristine C3N4 (blue) and CRed-AT-C3N4 (red). (b) Transient photocurrent. (c) Steady state and (d) transient state PL spectra.
催化性能的提高與構效關系
作者對系列樣品進行了光催化產氫性能測試(10%三乙醇胺-水體系)。如Fig. 6所示,與未做改性的C3N4相比,經Post-redox制備的CRed-AT-C3N4產氫活性提高了17倍,達到了12.3 mmol h-1 g-1 (300W Xe, 420nm-cutoff),420 nm處的量子效率為18.5 %。在550 nm以上仍能夠檢測到H2,說明光的利用效率較C3N4(~460 nm)有較大提高。通過分析各對照樣品,作者認為,從載流子分離的角度理解,超薄C3N4(AT-C3N4)和C還原C3N4(CRed-C3N4)分別對應于遷移距離的縮短和遷移速率的提高,二者都對催化性能有顯著的貢獻,而C還原的超薄C3N4產生了協同效應,催化活性便可進一步提升。
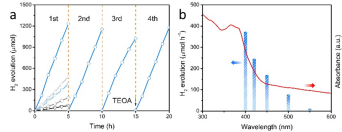
▲Fig. 6. (a) Time course of H2 evolution for pristine C3N4 (black), etched C3N4 (gray), CRed-C3N4 (light gray), AT-C3N4 (light blue) and CRed-AT-C3N4 (blue), (b) Wavelength-dependent efficiency of H2 evolution over CRed-AT-C3N4 (left axis) equipped with a serials of cut-off filters, respectively, UV-vis light absorption spectrum of CRed-AT-C3N4 (right axis).
結論
作者提出了一種同時對C3N4進行物理結構和電子結構調控的方法,制備出碳取代的超薄介孔C3N4納米片。這是一種簡單的自上而下的方法,理論上這種調控策略可通過改變輔助試劑來適用于不同的二維材料。這種催化劑既有利于傳質和載流子遷移,又能夠最大限度的利用可見光。






