

開篇語
原料藥雜質在USP中定義為任何不同于原料藥結構的物質,在中國藥典中為任何影響藥品純度的物質。ICH Q3A將原料藥中雜質分為有機雜質,無機雜質雜和殘留溶劑。原料藥中有機雜質來源于合成工藝相關的雜質及儲存過程中產生的雜質。有機雜質的控制對于藥物的安全性和有效性有著重要的意義。
一、關于法規
雜質研究與控制是藥物研究中的一項系統工程,CTD申報資料中與雜質相關的章節體現了雜質控制和終點控制相結合的策略,符合藥品質量研究中全面系統的質量控制理念。
原料藥申報資料中涉及到雜質研究與控制的內容有八個模塊小節:3.2.S.2.3生產信息中的物料控制,3.2.S.2.4關鍵步驟和中間體的控制,3.2.S.3.2特性鑒中的雜質譜分析,3.2.S.4.1原料藥質量控制中的質量標準,3.2.S.4.2分析方法,3.2.S.4.3分析方法驗證,3.2.S.4.4批檢驗報告和3.2.S.4.5質量標準制定依據等章節。
申報資料的撰寫應高度關注雜質分析與控制的系統性和法規對于雜質控制的要求。
ICHQ3A指南針對的是新原料藥中雜質的研究與控制,雜質的研究范圍是與工藝相關及原料藥降解的有機雜質。對于無機雜質、殘留溶劑和元素雜質的研究與控制在ICH中其它的相關指南有詳細敘述。
ICHQ3A指南中的雜質不包含原料藥中外來污染物質、多晶形物質及立體異構體。對于原料藥中基因毒性雜質的控制ICH Q3A不再適用,由ICH M7指南補充指導。
USP通則<1086, 原料藥和制劑雜質>的內容是原料藥和制劑中雜質的研究與控制指南。原料藥中雜質指的是與工藝相關及原料藥降解的有機雜質。
USP在2017年新建立了一個通則<476, 原料藥與制劑的雜質控制>,主要內容是藥典各論中已有的原料藥與制劑中雜質分析測試與控制,提出了有機雜質控制決策樹,如下圖,具體內容可參考PF43(6)。
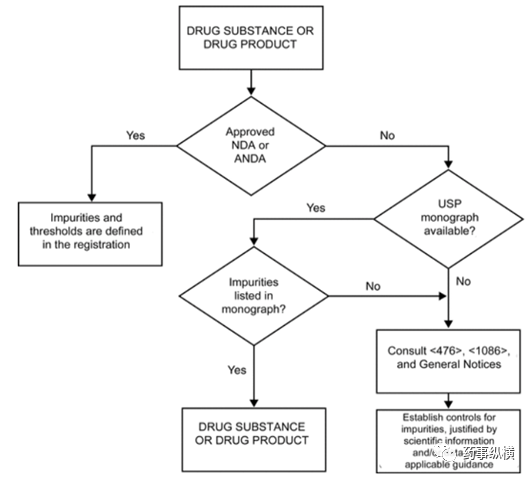
圖1:原料藥和制劑中有機雜質控制決策樹
二、關于原料藥不同階段的雜質控制
從藥品研發至產品上市通常需要經歷較長的時間,不同研發階段的關注點不同。各階段開展雜質研究的首要任務是患者的安全,根據擬定的治療適應癥、劑型、給藥途徑、給藥持續時間和患者人群,雜質研究根據具體情況,具體分析。
成本是階段性雜質研究的主要考慮因素之一。早期臨床研發階段需重點研究原料藥中最有可能存在和已經存在于的有機雜質。
原料藥合成過程中使用的起始物料、中間體、試劑、催化劑和溶劑均是明顯的潛在雜質。合成工藝中距離終產品原料藥的反應步驟是與潛在雜質被清除的可能性相關。最后一步反應的中間體、試劑和溶劑等,以及可預測的副產物和主要降解產物是這個階段雜質研究的重點。
在臨床III期及后面的階段,待商業化合成路線確定后,需要深入研究各步驟中潛在的雜質,開展雜質清除和衍生化研究,以確定合成工藝控制的關鍵點。隨著藥物臨床的進展,收集更多的批次雜質數據和穩定性數據,未知雜質的結構需要鑒定,必要時開發補充分析方以確認潛在雜質的存在與否。
對于遺傳毒性雜質,ICH M7指供了具體的指導。由于起始物料中的雜質可能影響到最終產品中的雜質控制,越接近最后產品合成步驟的起始物料,引入雜質的風險越高,因此對雜質研究和控制的要求也越高。起始物料中雜質控制策略在FDA與制藥企業在II 期會議中常討論的話題。ICH Q11指南闡述了起始物料控制,包括雜質控制及物料選擇的合理性。
工藝雜質的研究方式有“正推”和“逆推”兩種。“正推”就是從原料開始追蹤每一步反應,把每一步反應研究清楚,說明雜質的來源。“逆推”顧名思義是從產品開始向起始物料找到雜質的來源。“正推”方法能更清楚每一步反應的主副反應機理,對于雜質的來源會更加明確,從而優化工藝參數,確定關鍵工藝參數。
原料藥中的降解雜質是原料藥在儲存或穩定性研究過程中產生的雜質。降解雜質可以通過強制降解和影響因素實驗研究獲得。強制降解和影響因素條件與正常存放的情況有較大的不同,影響因素一般考察溫度、濕度、光度、空氣暴露等對產品質量的影響。
一、關于雜質研究中的分析方法技術
通常說的雜質譜包括藥物中雜質的種類、含量、來源及結構等信息。通過雜質譜分析能較為全面的掌握產品中雜質的概況。原料藥中雜質譜的研究需要有針對性地選擇合適的分析方法,確保雜質的有效檢出、定量和確認。
在新藥研究早期,會選擇與MS兼容的分析方法,有利于收集雜質的MS信息。后期根據需要調整適用于雜質屬性的方法。
對于藥典各論中雜質控制,通常通過標準品進行定量與定性;如果沒有標準品,則用RRT和RRF來進行定性和定量。藥典收載的一些雜質分析方法比較落后,專屬性較差。在進行仿制藥研究時需要更新這類分析方法,開發那些具有專屬性的色譜或光譜方法進行雜質控制研究。
面對雜質的微量和復雜性,檢測方法的專屬性、靈敏度和準確度十分關鍵。當原料藥中不同雜質不能同時被一種方法檢測時,應考慮開發不同分析方法,分別進行檢測。雜質分析方法的對象是各個潛在的雜質,因此分析方法的驗證需要根據不同雜質的特點綜合設計驗證方案,進行有針對性的規范驗證。
采用HPLC-UV/GM-FID方法可以滿足大部分雜質分析與控制的要求。對于微量和極微量的毒性雜質和基因毒性雜質,采用靈敏度更高的LC-MS/MS方法顯得尤為重要。采用MS技術可以對原料藥中的工藝雜質和降解雜質等成分進行結構特征的分析,有助于藥品中降解途徑規律的探討。
在整個藥品生命周期的雜質譜控制過程中,保證分析方法有良好的專屬性和敏感性顯得尤為重要。基于QbD理念開發分析方法,設計方案篩選關鍵影響因子,建立定量關系模型確定分析方法的操作空間,如下圖所示。
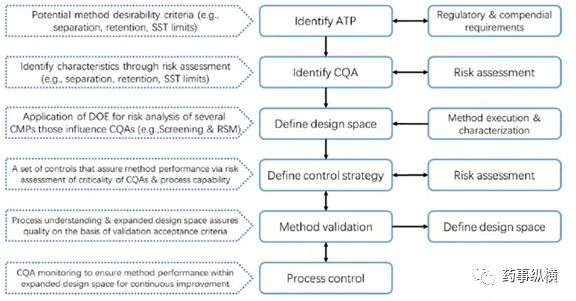
圖2:基于QbD理念的分析色譜方法開發
四、關于雜質限度制定
雜質的定量分析與分析方法的檢測限與定量限相關,因此雜質的分析方法需要經過驗證,包括檢測限與定量限的確定。
創新藥雜質限度的確定需要綜合藥物、藥理毒理和臨床研究結果綜合判定。在制訂質量標準中雜質限度時,安全性是首要考慮因素,其實是大生產和中試批次數據、穩定性數和工藝數據。如果雜質是藥物在人體中的主要代謝產物,則不考慮其安全性。創新藥研發早期,原料藥中設定的雜質限度不能高于毒理數據支持的限度水平,同時兼顧考慮生產的可行性和分析方法的波動性。
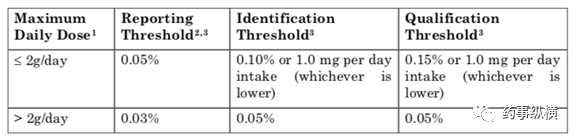
隨著臨床的研究進展,收集藥品的臨床前和臨床安全性數據以支持雜質限度的進一步優化。對于沒有毒理數據支持的原料藥雜質限度制定可以參考ICH Q3A指南,如表1。
表1:新原料藥中雜質的限度
臨床研究后期,應將商業化合成路線得到的放大生產樣品與臨床研究樣品中的雜質進行詳細比較,或已有雜質的含量超出原有的限度時,可參考ICH Q6A指南,按下圖3進行決策分析。基因毒性雜質的限度制定不在此文討論。
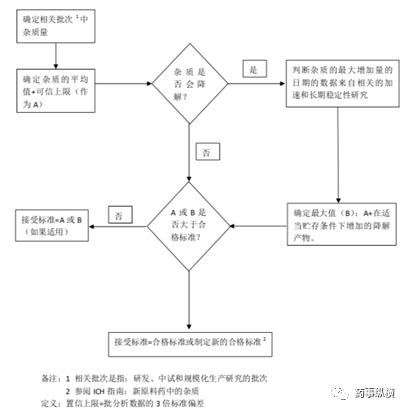
圖3:原料藥雜質限度制定決策樹
五、關于變更后雜質研究
對于工藝發生變更時,應重新進行雜質研究,評估現有分析方法是否適用。在藥物開發的早期,隨著藥物合成路線或工藝參數的變化,均可能出現新的雜質。若起始原料或中間體發生變更,需重新研究以確認它們和它們衍生的雜質,以及它們或它們衍生的雜質是否會殘留至原料藥中。溶劑和試劑發生變更也需將其作為新雜質進行研究。在進行工藝變更對雜質影響的風險評估過程中,需要檢測可能生成的新工藝雜質。這涉及到對潛在的新工藝雜質的預測能力、其被清除或轉移的可能性,以及現有的雜質檢測方法能否有效檢測它們。
藥物批準后的任何工藝方面的變更都應評估對雜質譜的潛在影響。除了工藝路線或所用材料的變更之外,還應包括一系列其他可能的變更。例如,生產場地、工藝控制點、生產規模和原料供應商等變更。
六、關于降解研究
大多數藥物的有效期并非受限于其有效性(即主藥含量),而是受限于其安全性(即生成的降解產物達到了關注閾值),因此藥物的降解研究在藥物研發階段非常重要。
監管機構鼓勵制藥公司在I/II期臨床研究階段報告強制降解實驗結果,期望通過原料藥的強制降解研究,保證藥物在前期臨床試驗期間的穩定性。根據獲得的降解雜質信息,初步了解藥物的降解途徑,期望在早期階段開發的方法具有穩定性指示性。
在III 期臨床研究階段,原料藥強制降解需要開展深入研究工作,以及對于藥物的降解途徑和降解方式進行深入的探討,獲得藥物在后期研發階段的潛在的穩定性問題,以確保產品貨架期質量可控。
降解實驗是預測、了解原料藥穩定性的重要工具。降解雜質可以通過強制降解和影響因素實驗研究獲得。強制降解實驗的目標是研究可能產生的和實際產生的降解產物,及用于開發和驗證具有穩定性指示性的分析方法。通過現代分析技術,收集降解雜質信息,對于研究原料藥降解途徑非常重要。
在過去十年的藥物降解研究中,Zeneth?軟件已成為計算機預測藥物理論降解途徑的有力工具。該軟件來自于Lhasa公司,它也是 METEOR? and DEREK?軟件的開發者。該軟件可以利用自身的藥物數據庫,減少預測的偏差,通過模型幫助藥物工作者預測分析和理解藥物的降解途徑。
另一個研究藥物降解研究的工具是Pharma D3數據庫。它由兩位化學家和ChemDraw 軟件的開發公司Cambridge 在2005年共同建立,數據庫收載了發表在文獻及公開研討會議中的藥物降解數據。研究者可通過藥物名稱、結構或者降解物的結構進行搜索,還可以分子量的變化進行搜索,以揭示特定官能團和藥物的降解模式。
結束語
本文討論了原料藥雜質研究中的一些思路和趨勢。仿制藥與創新藥雜質研究的思路不同,但殊途同歸,都需要關注各國法規和ICH指南的要求。創新藥研究在不同研究階段設定不同的目標,緊跟新技術的發展,利用工具快速有效的將研究工作推進。
藥物質量要考慮安全性、有效性和可控性。藥物的安全性常由藥物中雜質的含量決定,藥物的貨架期也常受加速和長期穩定性實驗中的雜質含量影響。降解研究是實現藥物雜質研究的有效的途徑之一,利用一些軟件工具可幫助分析工作者了解藥物的降解途徑和模式,幫助預測藥物的貨架期,提高藥物研發的效率,縮短研發周期,從面降低研發成本。






