

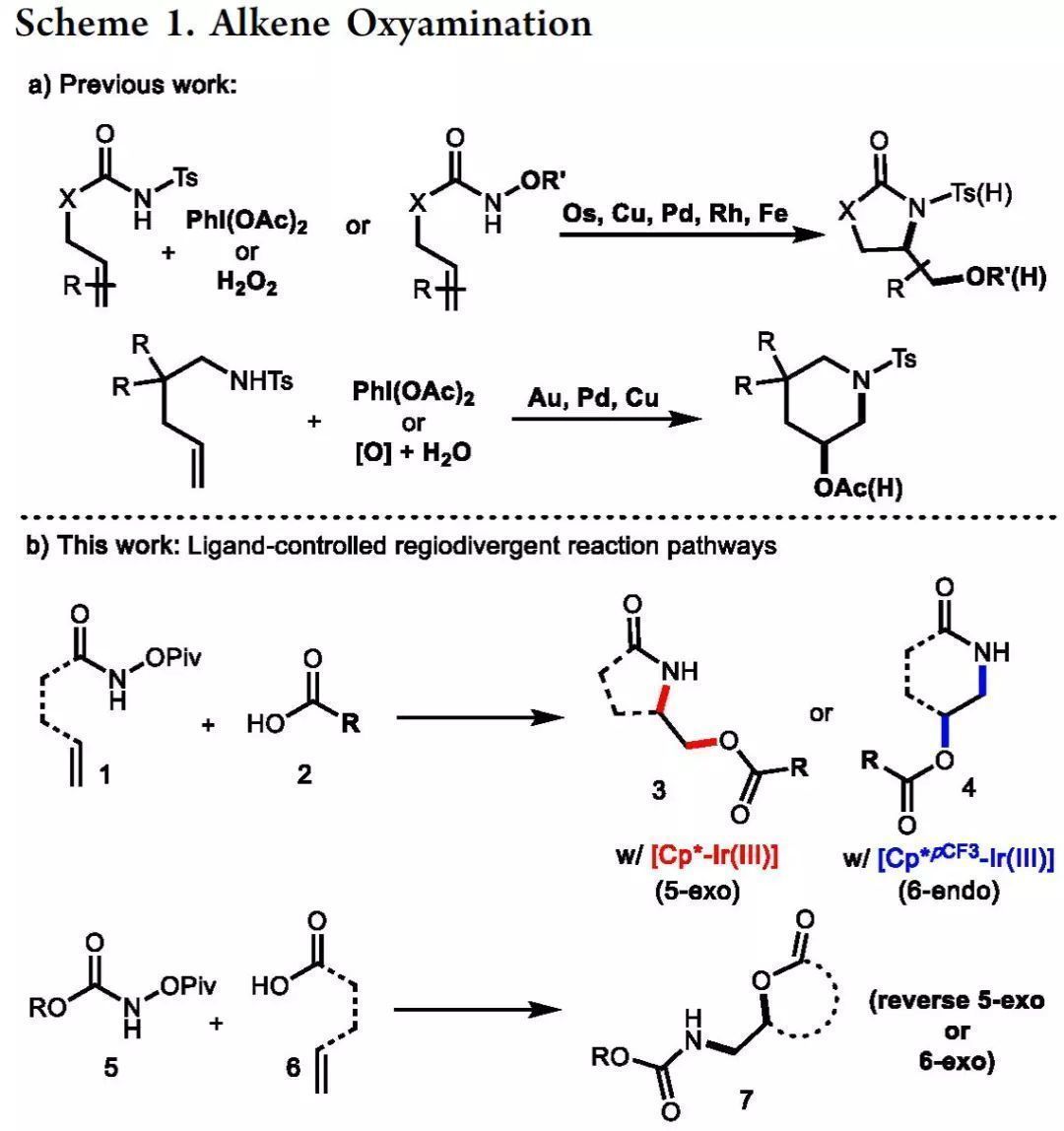
由于生物活性的天然產物和藥物中含有豐富的相鄰N, O結構和許多可用的烯烴前體,因此區域和立體選擇性的烯烴的羥胺化反應備受關注。受Sharpless開創的不對稱羥胺化反應啟發,目前已經發展了涉及Os, Cu, Pd, Rh, Fe, Au和Mn等過渡金屬催化以及其他策略(Scheme 1a)。盡管已取得重要進展,但烯烴的區域選擇性羥胺化仍具挑戰。近日,美國科羅拉多州立大學Tomislav Rovis教授課題組報道了由Ir上Cp配體調控的未活化烯烴的區域選擇性羥胺化反應,更缺電子體系有利于6-endo選擇性,而Cp*則具有高的5-exo選擇性。此外,烯基酸和外源性亞硝基前體的反應則會使區域選擇性翻轉(Scheme 1b)。相關研究成果發表在J. Am. Chem. Soc.上(DOI: 10.1021/jacs.9b06366)。
(來源:J. Am. Chem. Soc.)
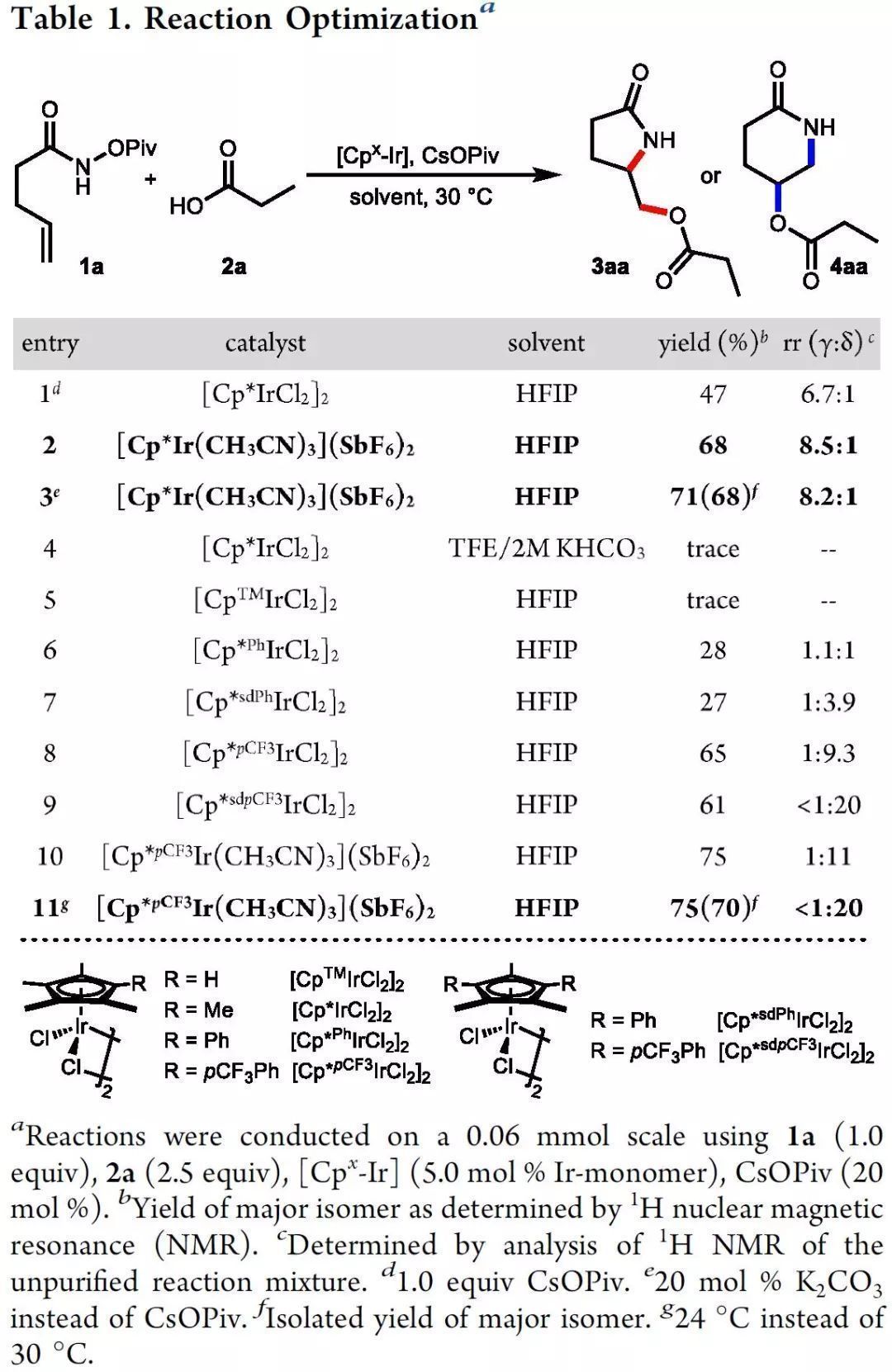
研究初期,作者以連有N-新戊酰異羥肟酸酯的烯烴1a與丙酸2a為模型底物,考察了該偶聯反應的可行性。作者高興地發現在HFIP中,[Cp*IrCl2]2與CsOPiv的組合能促使反應以47%的收率和6.7:1的區域選擇性得到目標羥胺化產物3aa,且更傾向于形成γ-內酰胺(5-exo)。預制的陽離子銥催化劑可以避免氨氯化副產物的形成,收率進一步提高至68%(entries 1-3)。隨后作者考察了更多缺電子的Cp配體,它們能明顯提高δ-內酰胺4aa的反應性和選擇性(entries 5-9)。相應的陽離子銥復合物效果最佳,且在室溫下反應時,產物的選擇性最高(entry 11)。
(來源:J. Am. Chem. Soc.)
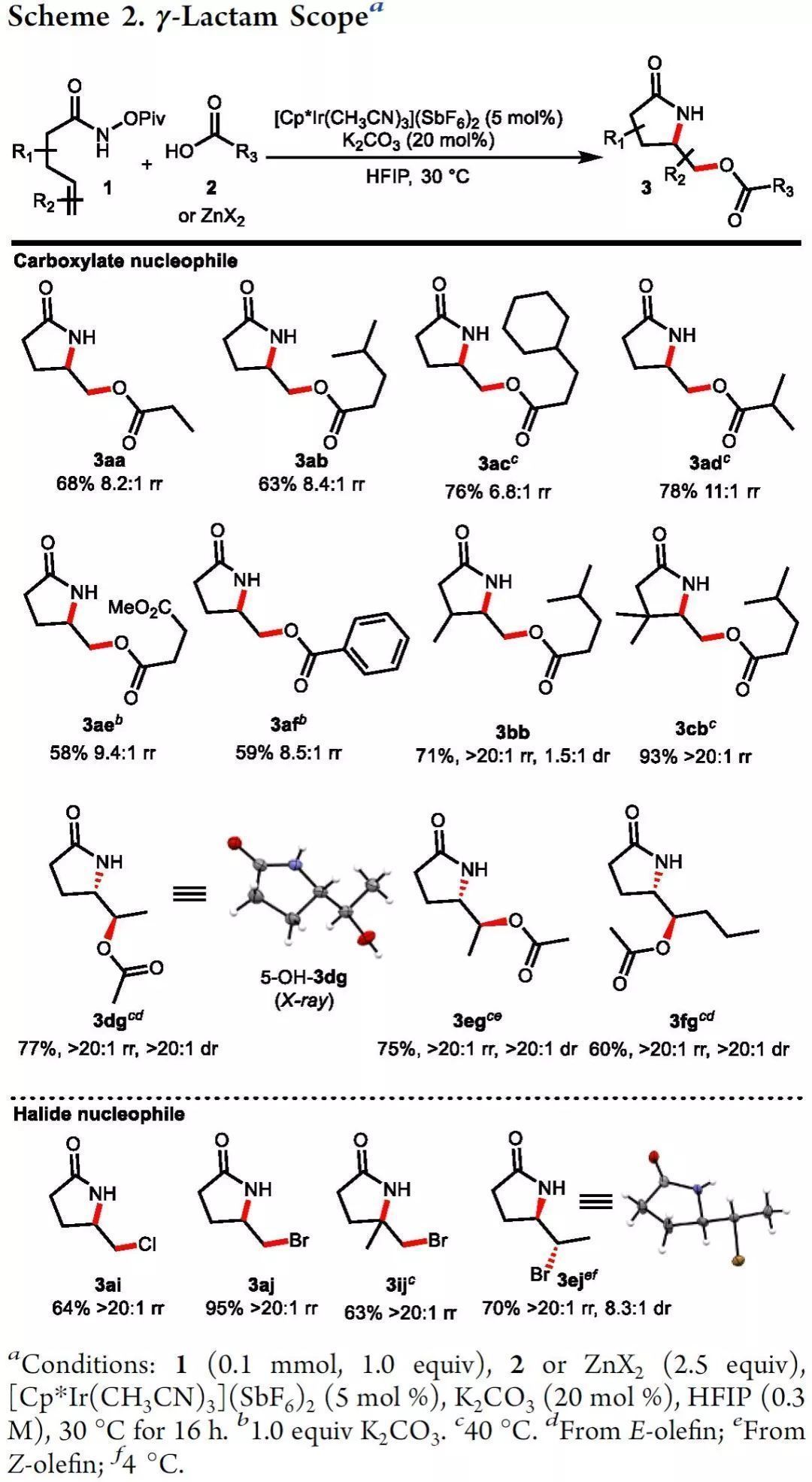
接著,在最優反應條件下,作者首先考察了形成γ-內酰胺(5-exo)的適用范圍(Scheme 2)。該反應可耐受多種脂肪酸和芳香酸(3aa-3af)。β-位帶有取代基的鏈烯基異羥肟酸酯可以順利得到相應的產物(3bb、3cb),收率較高。更重要的是,含有內烯烴的底物也是相容的,順式和反式1,2-二烷基烯烴形成了互補的非對映體(3dg-3fg),這表明氮氧基團與雙鍵是立體選擇性的反式加成反應。
為進一步拓展此方法,作者還考察了氯化物和溴化物等親核試劑。經條件篩查,作者發現鹵化鋅是最佳的鹵化物源,能以良好的收率提供氨氯化和溴化產物(3ai、3aj、3ij、3ej)。
(來源:J. Am. Chem. Soc.)
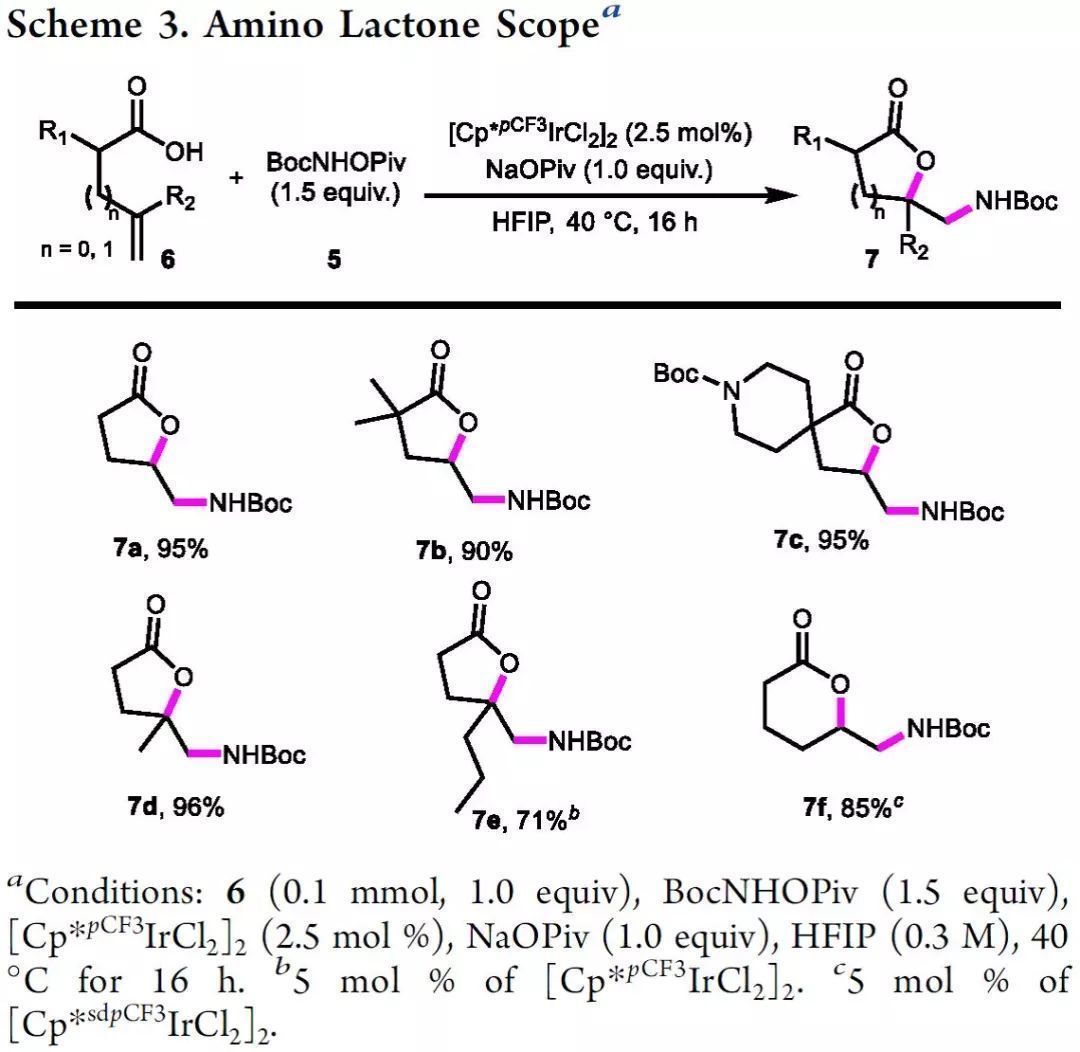
由于1a能區域選擇性地得到氨基醇,作者設想烯酸能否通過類似的機理與外源的異羥肟酸酯結合(Scheme 3)。令人高興地是,當以缺電子[Cp*pCF3IrCl2]2作催化劑時,反應順利進行,并以幾乎定量的收率得到所需的氨基內酯(7a-7e)。更重要的是,利用更缺電子的Cp*sdpCF3Ir(III)催化劑也可以高收率實現6-exo環化過程。
(來源:J. Am. Chem. Soc.)
接下來,作者考察了δ-內酰胺(6-exo選擇性)的適用范圍(Scheme 4)。該反應兼容多種脂肪族和芳香酸(4aa-4ao),并且可以很好地耐受不同的取代基,如β-鹵化物或α-雜原子等。此外,與γ-內酰胺的合成不同,α-取代底物能以優異的非對映選擇性轉化為δ-內酰胺(4jb,4kb)。

(來源:J. Am. Chem. Soc.)
最后,基于機理實驗和以往文獻報道,作者提出該羥胺化反應的合理機制。γ-和δ-內酰胺的發散性合成取決于Cp配體的電子性質。當使用富電子的Cp*時,通過Ir(V)-氮賓中間體形成γ-內酰胺。相反,當利用缺電子的Cp*pCF3時,在形成Ir-氮賓之前,羧酸負離子先對活化的烯烴發生親核進攻進而形成δ內酰胺(D)。最后,區域互補的氨基內酯產物是由羧酸鹽捕獲活化的Ir-氮賓烯烴復合物產生的(F)。

(來源:J. Am. Chem. Soc.)
總之,作者發展了未活化烯烴的區域發散性和立體選擇性的羥胺化反應。區域選擇性是由Cp配體的電子性質決定。相關γ-內酰胺、γ-內酯和δ-內酰胺等也可以由簡單獲得的原料快速組裝得到。






