

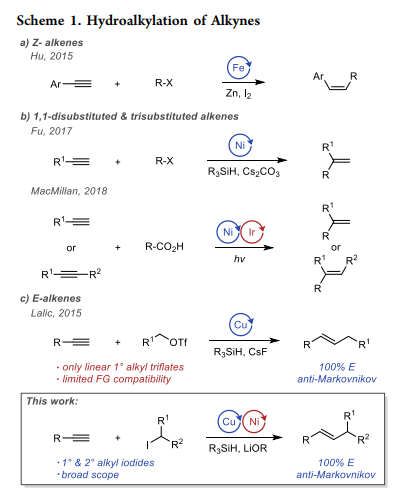
炔烴的加氫烷基化最近已成為烯烴合成的有效新方法。Z-取代的芳基烯烴可以使用芳基炔烴的自由基加氫烷基化制備(Scheme 1a),Fu和MacMillan小組分別開發了鎳催化和光氧化還原/鎳協同催化的末端炔烴向1,1-二取代烯烴的轉化方法(Scheme 1b)。然而,能夠有效的合成二取代E-烯烴的加氫烷基化方法卻很少報道。2015年,Gojko Lalic小組報道了使用烷基三氟甲磺酸酯作為親電試劑的銅催化末端炔烴的加氫烷基化(Scheme 1c),為合成E-烯烴提供了有效方法。但該方法具有一些明顯的局限性,比如對親電試劑的空間性質高度敏感,只適用于沒有α-支鏈的直鏈伯烷基三氟甲磺酸酯。另外,烷基三氟甲磺酸酯和試劑的高反應性進一步限制了反應范圍,并引入了與起始親電試劑的制備、純化和穩定性相關的實際問題。
(來源:J. Am. Chem. Soc.)
近日,美國華盛頓大學Gojko Lalic小組在此基礎上開發了一種新的末端炔烴加氫烷基化方法,解決了之前原始加氫烷基化反應的關鍵缺點,同時保留了其主要優點。證明Cu/Ni雙催化劑體系允許使用伯和仲烷基碘作為偶聯配偶體并且能夠從末端炔烴立體特異性地合成寬范圍的E-烯烴。相關研究成果發表在J. Am. Chem. Soc.上(DOI: 10.1021/jacs.9b04800)。
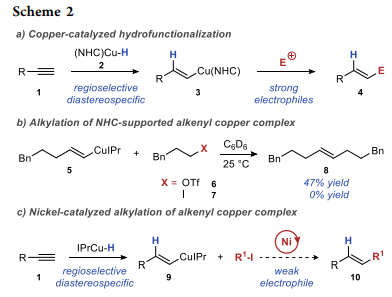
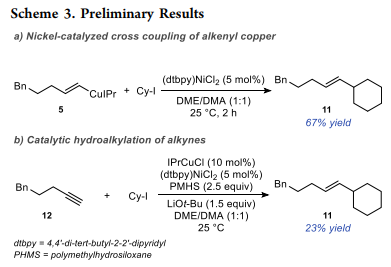
受Nakao和Brown的開創性工作的啟發,作者計劃使用鎳助催化劑促進有機銅中間體與烷基碘的交叉偶聯(Scheme 2c)。初始實驗中,作者發現常見的交叉偶聯催化劑(dtbpy)NiCl2促進了分離的鏈烯基銅中間體5和環己基碘的有效交叉偶聯(Scheme 3a)。然而,在催化加氫烷基化反應中,相同的鎳催化劑僅以23%的收率得到所需的E-烯烴(Scheme 3b),與各種單和雙齒膦和氮基配體的反應同樣不成功。
(來源:J. Am. Chem. Soc.)
與作者預期相反,馬氏加氫烷基化產物在這些反應中不是重要的副產物,相反,主要問題是形成了高分子量產物的混合物。作者推斷高分子量的副產物可能是通過Reppe的三聚化和鎳(0)絡合物促進的炔烴的相關四聚化形成的。考慮到Reppe三聚化的機理,三齒配體應通過防止兩個炔烴分子同時配位來抑制這種副反應。作者發現,使用IPrCuCl作為催化劑,三齒tpy配體負載的鎳(I)作為助催化劑,Ph3SiH用作氫化物源,LiOi-Pr用作銅催化劑的轉換試劑能夠使末端炔烴12和環己基碘化物反應得到最佳結果(Table 1)。

(來源:J. Am. Chem. Soc.)
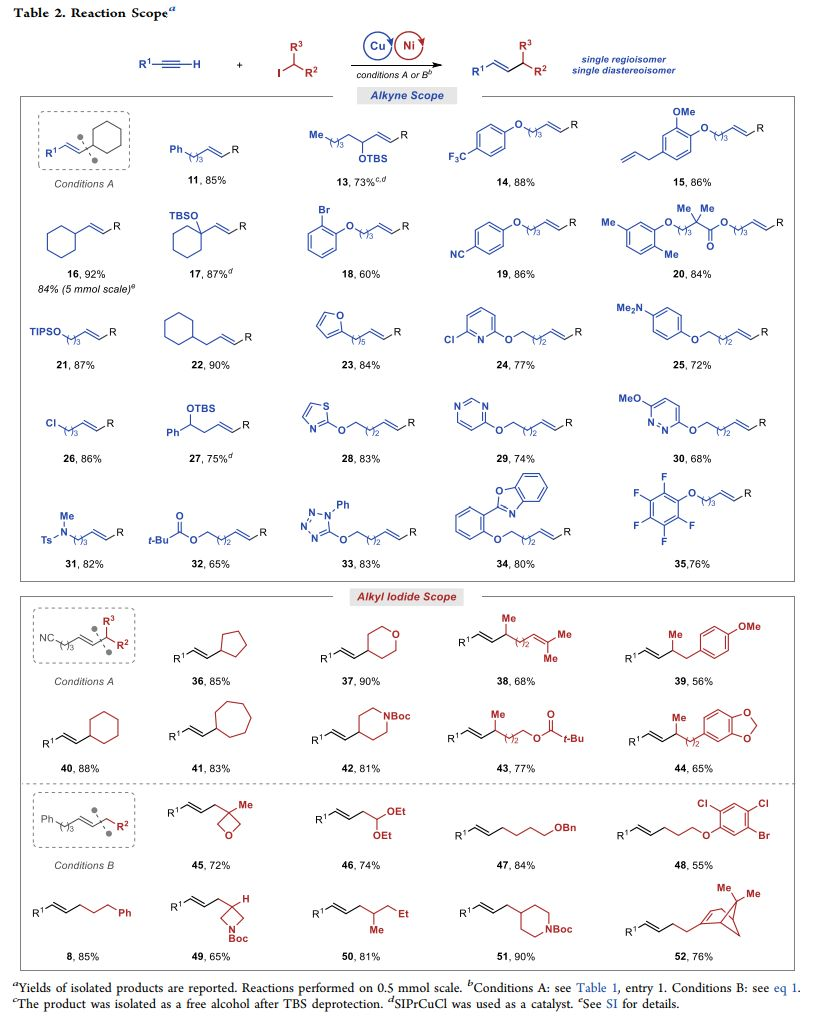
進一步,作者研究了該反應的范圍。如Table 2所示,所有產物都是作為單一區域異構體和單一非對映異構體獲得的。該反應具有優異的官能團容忍性,包括腈類(19)、酯(20)、芳基醚(15)、甲硅烷基醚(13,17,21和27)、烯烴(15)、烷基氯(26)、磺胺類(31)、二烷基苯胺(25)和芳基溴化物(18)。新反應還可耐受多種雜芳烴,包括呋喃(23)、2-氯吡啶(24)、噠嗪(30)、噻唑(28)、嘧啶(29)、四唑(33)和苯并惡唑(34)。但反應不能容忍諸如羥基和氨基的質子官能團,可還原的官能團例如醛和活化的烯烴也與反應條件不相容。叔烷基碘化物、芳基取代的炔烴和二取代的炔烴也不參與加氫烷基化反應。
(來源:J. Am. Chem. Soc.)
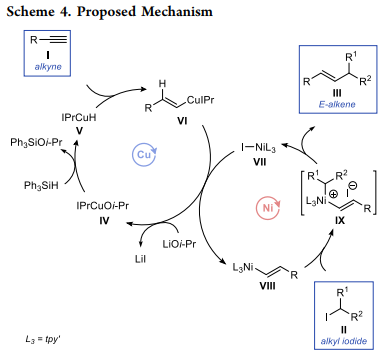
如Scheme 4所示,基于銅催化的炔烴加氫官能化和鎳催化的交叉偶聯反應的典型機制,作者提出了加氫烷基化反應的機理。
(來源:J. Am. Chem. Soc.)
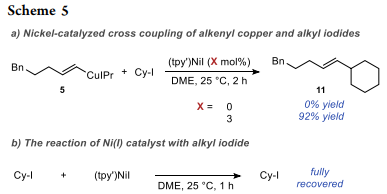
進一步,作者通過實驗證明了所提出的鎳催化的鏈烯基銅中間體和烷基碘交叉偶聯的可行性(Scheme 5)。最后,作者探討了金屬轉移反應和烷基鹵化物活化之間的關系。結果表明,在該反應中,金屬轉移(VI→VIII)可能發生在烷基碘活化之前(VIII→IX)。
(來源:J. Am. Chem. Soc.)
結語:美國華盛頓大學Gojko Lalic等人開發了一種從末端炔烴和烷基碘化物立體特異性的合成E-烯烴的方法。加氫烷基化反應通過銅和鎳的協同催化作用實現,并且具有優異的反馬氏加成選擇性。反應具有廣泛的范圍。機理研究提供了銅催化劑通過加氫活化炔烴的證據,是控制整個反應的區域選擇性和非對映選擇性的關鍵。






