烷基氯化物是化學合成中常用的起始原料,它穩定性好且價格低廉。在各種親核取代和芳烴取代反應中,它們是經典的烷基化試劑和親電試劑。然而,烷基氯化物也有其局限性,由于C-Cl鍵鍵能高(327 kJ/mol),與C-Br鍵(285 kJ /mol)和C-I鍵(213 kJ/mol)相比,它的反應活性要低得多。在對其進行還原活化時,烷基氯化物需要較長的反應時間和特別苛刻的反應條件,往往會導致非選擇性轉化。近年來,光誘導單電子轉移已成為活化烷基C-X鍵的有力工具,它能夠使烷基鹵化物形成具有親核性的碳自由基,這類自由基是構建C(sp3)-C鍵和C(sp3)-H鍵的重要中間體。與離子類型的反應相比,自由基反應有許多優點,例如對空間位阻相對不敏感,對不飽和碳-碳鍵加成后形成的過渡態允許生成熱力學不穩定的產物。
絕大多數烷基氯化物的還原脫鹵僅限于一些活化的底物,如芐基氯、α-羰基氯化物或含有三氯甲基的化合物,而對未活化氯化物的脫氯反應仍然面臨著巨大的挑戰(圖1a)。2018年,Matsubara及其研究團隊報道了在UVA輻射誘導的光催化劑雙(二甲氨基)-咔唑的條件下,實現了某些烷基氯化物的脫氯反應(J. Org. Chem., 2018, 83, 9381–9390)。隨后,Claros和Lloret-Fillol等人采用鎳和鈷絡合物的雙重催化作用來活化烷基氯并誘導其還原環化(Angew. Chem. Int. Ed., 2019, 58, 4869–4874)。Goez、Kerzig和Wenger等人則報道了通過在水溶液中生成溶劑化電子或三重態-三重態湮沒上轉換來活化各種水溶性氯化物,實現C-Cl鍵的光催化活化(Chem. Sci., 2016, 7, 3862–3868; Chem. Sci., 2018, 9, 6670–6678)。
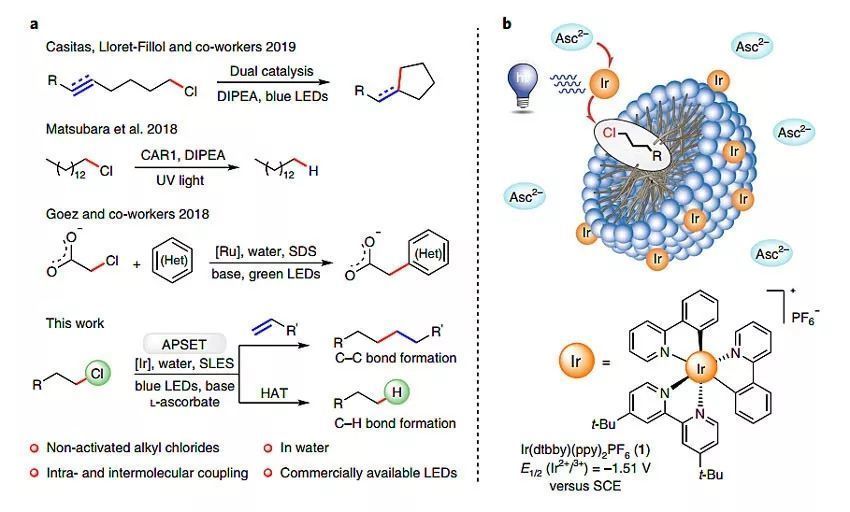
圖1. 光驅動的C-Cl鍵的活化。圖片來源:Nat. Catal.
光氧化還原催化已成為合成各種有機化合物的有力工具。最近,德國雷根斯堡大學的Burkhard K?nig教授課題組利用了一種組裝促進的單電子轉移(APSET)策略,使未活化的烷基氯化物產生碳自由基,并將其應用于脫氯、加成和環化反應(圖1)。這種新開發的方法操作簡單,利用標準、廉價的市售藍色LEDs即可實現。機理研究表明在一個催化循環中有兩個可見光光子的能量累積。相關結果發表在近期的Nature Catalysis 期刊上。
根據文獻的報道,作者認為分隔反應物的微膠束溶液可能會穩定活性較強的銥絡合物中間體。在水溶性電子供體和恒定藍光照射下,于十二烷基低聚乙二醇硫酸鈉(SLES)中對Ir(dtbby)(ppy)2PF6(1)的紫外可見吸收光譜進行了測試,它在533 nm處出現的特征光譜變化表明了[Ir(dtbby)?-(ppy)2]的形成(圖2),與常用的有機溶劑和胺作為電子給體的體系相比,水/SLES混合物可以穩定[Ir(dtbby)?-(ppy)2],它在溶液中的穩定存在增加了對其進行連續激發的可能,或許能夠對穩定的烷基氯化物進行還原。此外,這些膠束不僅能使烷基氯化物溶解,而且在親水的氯原子指向膠束表面時,在空間中形成有利的預聚集,進而會促進單電子轉移到底物上從而產生自由基陰離子。
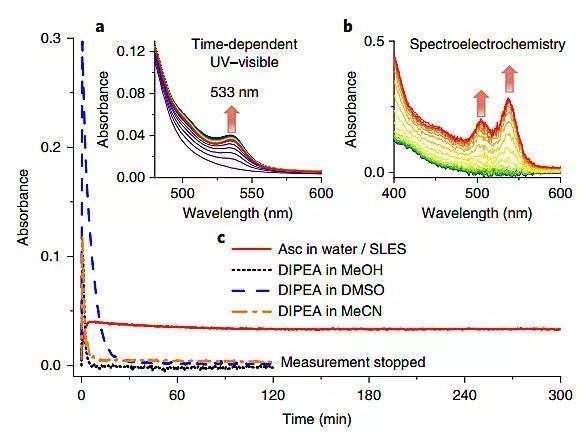
圖2. [Ir(dtbby)?-(ppy)2]的光譜測試。圖片來源:Nat. Catal.
為了驗證烷基氯化物在設計的體系中優先被還原的假設,作者將(3-氯丙基)苯(2a)作為反應的底物,Ir(dtbby)(ppy)2PF6(1)作為光催化劑,SLES和L-抗壞血酸水溶液作為還原劑。在優化的反應條件下,21 h后2a的轉化率為76%,并且以60%的收率得到還原脫氯產物2b。控制實驗表明,光照、光催化劑和SLES都是反應必不可少的條件;作為電子供體的抗壞血酸同樣也是不可或缺的。反應時,肉眼能夠觀察到2a的充分溶解,動態光散射(DLS)實驗也進一步證實了這一點。相反,在沒有1-戊醇作為助表面活性劑的情況下,反應會產生渾濁的混合物,烷基氯化物的溶解度有限,從而大大降低了收率。先前報道表明在有機溶劑條件下并不能有效地還原烷基溴化物,這進一步證明了微非均相環境對此反應的重要性。
一系列非活化的氯化物在優化的條件下通過還原脫氯反應,以中等到優秀的收率得到了相應的烷烴(圖3)。為了對反應的選擇性進行評價,作者考察了底物2a-13a的轉化率以及脫氯產物2b-13b的產率,其中一級和二級脂肪族氯化物(2a-4a、6a、9a、11a和12a)均以高收率和高選擇性得到了脫鹵還原產物;對于三級氯化物13a,除了還原脫氯產物外,還伴隨有通過SN1反應生成的叔醇副產物。盡管該方法在強堿性反應條件下進行,但它對苯甲醚(4a,67%)、雜芳環(6a,75%)、縮醛(9a,77%)、酰胺(11a,66%)、羧基(5a,42%)以及羥基(10a,32%)等官能團均耐受;除了烷基氯化物外,該方法也可以實現氯苯的脫氯還原(7a,47%)。
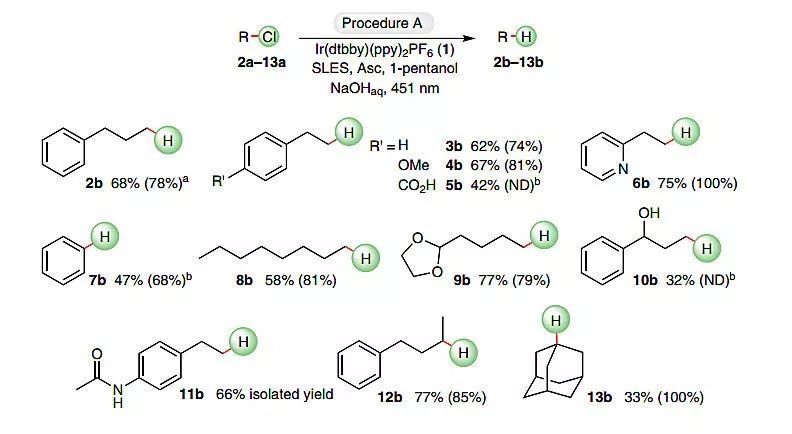
圖3. 烷基氯脫鹵還原反應中底物的范圍。圖片來源:Nat. Catal.
此外,作者設想將烷基氯化物的活化應用于分子間C-C鍵的形成。以碳為中心的烷基自由基具有親核性,可以與親電試劑發生反應。因此,作者選擇了缺電子的烯烴與其進行Giese型偶聯反應。與脫鹵方案相比,烯烴的加入明顯影響了反應活性和微結構溶液的組成。因此,以丙烯腈(14)為底物,對反應條件進行了優化,結果表明相對較高的pH值提供了所需濃度的L-抗壞血酸二價陰離子,反應在451 nm和400 nm兩種LEDs照射下進行,而在綠光下不進行,同時更高的光照強度會加速反應的進行。
由于伯醇類助表面活性劑具有一定的親核性,可能會與缺電子的烯烴發生副反應;助表面活性劑在界面上與極性烯烴可能會存在競爭。為了克服這些問題,作者認為選擇適當的烯烴可以作為助表面活性劑并適合表面活性劑層結構,從而增加界面膜的柔韌性,DLS表明丙烯腈(14)具有助表面活性劑的特征(圖4)。
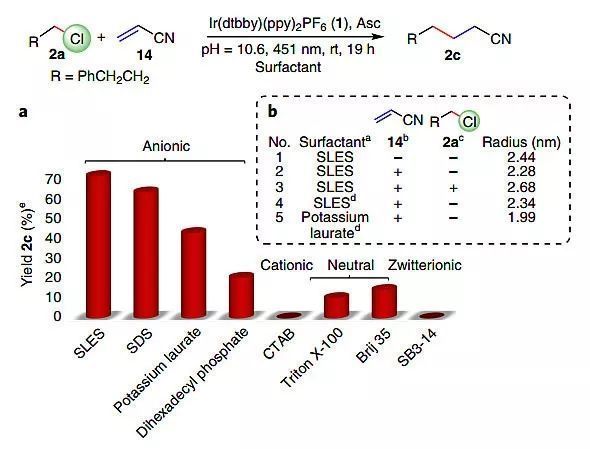
圖4. 烷基氯脫鹵形成C-C鍵。圖片來源:Nat. Catal.
為了更好地理解納米結構對催化反應轉化率和產率的影響,作者對表面活性劑的類型進行了研究(圖4),陽離子和兩性離子表面活性劑能夠形成微非均相體系,但反應產率為零。中性表面活性劑Triton X-100和Brij-35的產率很低,僅為10%和14%。相反,陰離子表面活性劑更利于反應,硫酸根基團比羧酸根基團更有效,因此硫酸鹽與[Ir(dtbby)?-(ppy)2]的相互作用要比羧酸鹽強得多。
在優化的條件下,作者考察了其它烯烴與(3-氯丙基)苯2a的反應(圖5)。氰基α-位甲基的存在進一步促進了2d的生成,而β-位帶有取代基則抑制了2e的形成。正如預期的那樣,疏水性更強的烯烴17與2a反應時,只有微量的偶聯產物2f生成。此外,在優化的條件下以甲基丙烯腈(15)作為自由基受體對烷基氯化物進行了拓展,一級氯化物和二級氯化物都能以較高的收率得到相應的產物2d和12d,叔氯只有少量的自由基偶聯產物18d生成,可能是由于底物18a的快速水解引起的。盡管甲氧基4a、吡啶環6a或酰胺11a可能通過氫鍵與催化體系的水相相互作用,但它們仍然具有良好的耐受性。
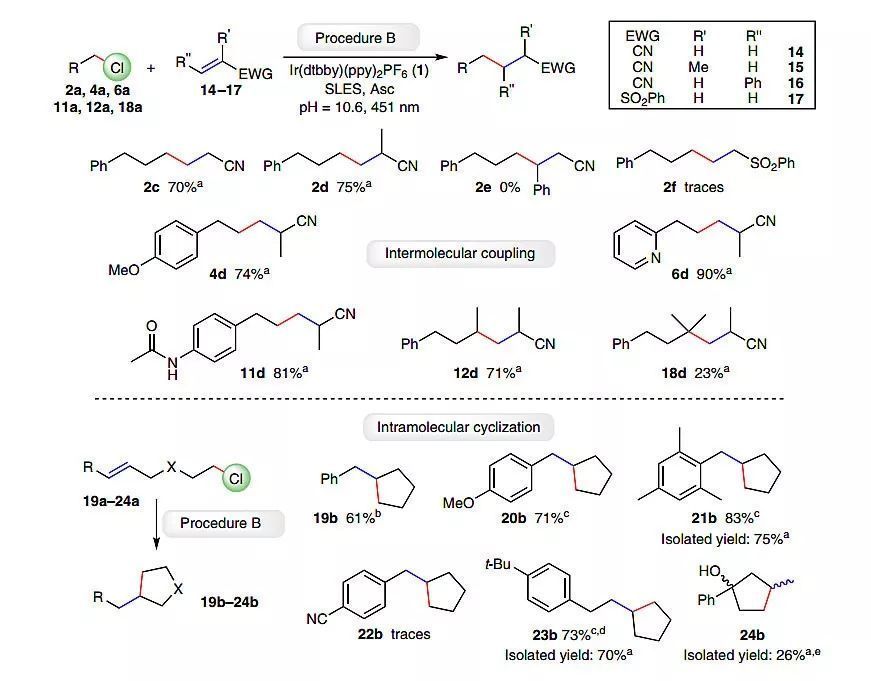
圖5. 烷基氯脫鹵偶聯反應中底物的范圍。圖片來源:Nat. Catal.
自由基環化反應由于能夠構建復雜的生物分子而受到廣泛關注,作者以苯乙烯衍生物19a-21a進行了簡單的5-exo-trig環化反應(圖5),均能以較高的收率得到相應的環戊烷產物19b-21b。除非取代基促進副反應,否則與雙鍵相連的芳環中電子和位阻效應對環化反應的結果幾乎沒有影響。然而,含有末端雙鍵的親水底物(24a)的反應活性較低,可能是由于它干擾了膠束表面的預聚集,當加入助表面活性劑1-戊醇時,能夠得到產物24b。
為了確定所研究的APSET系統涉及一個還是兩個光子的整體連續吸收,作者將Ir(dtbby)(ppy)2PF6還原為[Ir(dtbby)?-(ppy)2],并在嚴格的氬氣氛圍下加入氯化物2a,避光反應19小時,發現底物2a沒有發生轉化。這一結果表明,處于基態的[Ir(dtbby)?-(ppy)2]不能活化烷基氯化物2a,也說明了反應中的催化活性源于兩個光子的連續吸收,從而產生具有足夠還原能力的中間體(圖6a)。
在分子內的偶聯反應中,烷基氯和烯烴都可能被還原,為了對這一過程進行研究,作者設計了氯化物25a的自由基鐘反應(圖6b),主要產物環戊基丁腈25b的生成表明25a生成了自由基,經過5-exo-trig環化反應后被丙烯腈14捕獲。此外,作者還進行了氘標記實驗(圖6c),反應以氘水為溶劑,它既是很好的氘陽離子來源,又是弱氘原子的供體。氯化物4a的脫鹵反應得到了4b和4c的混合物,其中4c含有52%的氘。由于β-位的自由基反應活性高且不穩定,因此C–D鍵的形成可能是通過DAsc物種的氘原子轉移(DAT)機制,其在反應混合物中原位形成。其余48%的氫來自于具有長烷基鏈的(共)表面活性劑。與4a相比,氯化物2a和20a生成的自由基具有更高的穩定性和更低的負還原電位,使得它們更容易被二次還原,得到完全氘代的產物2g和20c。然而,在這種情況下,也無法排除存在部分DAT途徑。Ir(dtbby)(ppy)2PF6在混合膠束中的光譜與在DMSO中相比,具有更短的熒光壽命和更低的發光強度,并且最大紅移為23 nm。

圖6. 機理研究。圖片來源:Nat. Catal.
最后,作者提出了該反應可能的機理(圖7)。所有實驗都證實了微非均相環境在光活化和自由基反應步驟中的決定性作用,陽離子銥絡合物1以非共價鍵的形式固定在膠束的負電荷表面。當它被藍光照射激發后,抗壞血酸二價陰離子對其進行還原,生成的[Ir(dtbby)?-(ppy)2](E1/2= -1.51 V vs SCE)不足以誘導電子轉移到烷基氯(E1/2= -2.80 V vs SCE),隨后被第二個光子繼續激發,生成[Ir(dtbby)?-(ppy)2]*。無論是通過外層的電子轉移還是通過溶劑化電子的中間作用,烷基氯被還原后,生成自由基陰離子E,由于氯化物陰離子的水化能較高(320 kJ/mol),水溶液的環境促進了C–Cl鍵的裂解。自由基F易于從(共)表面活性劑或抗壞血酸單陰離子中攫取氫原子形成脫鹵產物,或與烯烴進行偶聯反應。緊密靠近界面處的缺電子烯烴進一步促進了分子間偶聯反應的進行。
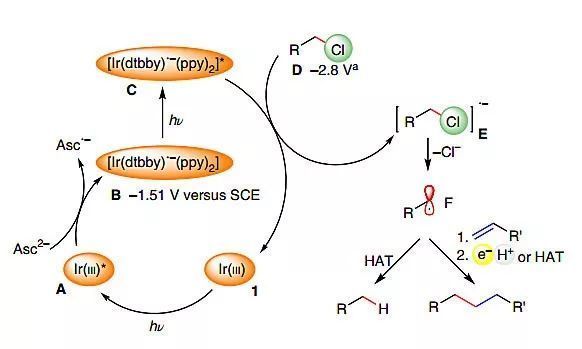
圖7. 可能的機理。圖片來源:Nat. Catal.
Burkhard K?nig課題組通過APSET系統在可見光誘導條件下對高還原電位的烷基氯化物進行了活化。水膠束溶液通過非共價鍵的相互作用和反應物質的隔離來穩定[Ir(dtbby)?-(ppy)2]絡合物,這種方法在烷基氯化物脫鹵、加成和環化反應中具有潛在的應用價值。機理研究表明,該反應以conPET的方式利用了兩個光子的能量。該結果為非活性氯化物作為烷基自由基源在有機合成中的應用開辟了新的道路。








