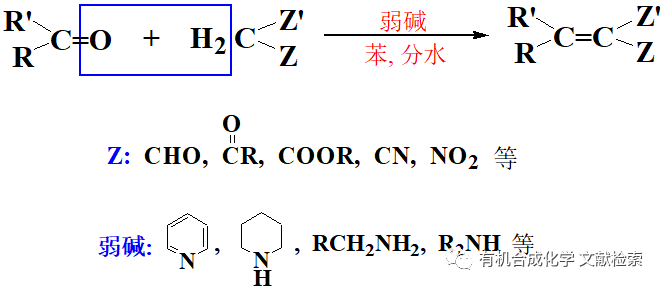
通過過渡{attr}2126{/attr}催化的碳-碳σ鍵活化構建新的碳-碳/雜鍵是有機化學中的主要研究課題之一。由于張力釋放驅動力,三/四元飽和/不飽和碳環是該類轉化的理想候選物。因此,擁有高張力的亞甲基環丙烷(MCPs)的反應性引起人們的廣泛關注,其可能反應模式包括近端外式亞甲基雙鍵的活化(C1-C2活化)和通過遠端單鍵C2-C3或C3-C4插入形成金屬環丁烷化物(Scheme 1a)。盡管存在區域選擇性問題,但涉及C3-C4裂解的MCP的加氫官能團化將提供一種獲得各種末端烯烴的有效方法(Scheme 1b)。
此外,末端烯烴是有機合成的重要中間體,可以用于聚合、復分解、環氧化、加氫甲酰化、加氫胺化等反應中。因此,需要開發簡單可靠的方法獲得通用的末端烯烴。為此,Yamamoto、施敏和Mascarenas等人通過選擇性C-C遠端鍵斷裂與親核性較強的前親核體如氮、氧和碳負離子親核試劑進行MCP的加氫官能團化,制備得到各種官能團化的末端烯烴。然而,上述方法均未能獲得烷基取代的末端烯烴。因此,開發通過簡單的烷基試劑對MCP進行加氫烷基化的方法具有重要意義。近日,加拿大麥吉爾大學李朝軍課題組利用簡單的腙作為碳負離子等價物,通過高選擇性的碳-碳σ鍵斷裂實現了鈀催化的亞甲基環丙烷與簡單腙的加氫烷基化(Scheme 1c)。該成果發表于近期Chem. Sci.(DOI: 10.1039/D0SC01221A)。

(圖片來源:Chem. Sci.)
首先,作者考察了以MCP 1a與苯基腙2a為模型底物進行了研究(Table 1)。在[Pd(allyl)Cl]2(5 mol%)、IPr·HCl(20 mol%)和1.2 eq. KOH的THF溶液中,1a與2a反應得到加氫烷基化產物(收率80%)。不存在鈀或堿時反應未能發生;其他鈀催化劑(如Pd2(dba)3)效率較低;NaOH與KOH效果類似,而其他堿反應性較差。配體在反應中起重要作用:NHC配體效果較好,而其他配體如PCy3和dppp效果較差。1,4-二氧六環為最佳反應溶劑。升高和降低溫度均導致收率降低。

(圖片來源:Chem. Sci.)
確定最佳反應條件后,作者考察了腙的底物范圍(Table 2)。含有甲基、甲氧基、氟、三氟甲基和N,N-二甲胺基等官能團的腙、由多環芳香醛(如1-萘醛)衍生的腙以及對位、間位、鄰位和多取代的芳香腙均適用該反應。此外,該反應還適用于由含呋喃、吡咯和吲哚的雜芳基醛制備的腙以及脂肪醛衍生的腙。

(圖片來源:Chem. Sci.)
接下來,作者考察了亞甲基環丙烷(MCP)的底物范圍(Table 3)。一般情況下,MCP均能順利反應并以中等至良好的收率得到加氫烷基化產物。在最佳反應條件下,芳環上可以耐受甲基、甲氧基、氟等各種官能團。此外,含炔基的底物具有良好的耐受性。除在R’位含有甲基和苯基的底物外,其他烷基取代的MCP以及環狀MCP也適用于該反應。然而,當R’為H時,得到3na和3na’(3:2)的區域異構體混合物。

(圖片來源:Chem. Sci.)
為了深入了解該轉化,作者進行了初步的氘標記實驗(Scheme 2)。當2a-d2與1b反應時,得到氘代產物3ba-d2,其中氘代發生在C1(52% D)和C4(78% D)位。

(圖片來源:Chem. Sci.)
基于上述實驗結果和前期研究,作者提出了可能的反應機理(Scheme 3)。首先,預催化劑[Pd(allyl)Cl]2經還原產生Pd(0);隨后,Pd(0)直接插入MCP 1的遠端σ鍵中得到Pd-環丁烷I;堿促進腙2與Pd-環丁烷I的反應形成中間體II;然后,中間體II生成π-allyl-Pd III或III’;最后,III分解釋放出N2并生成產物3完成催化循環(Scheme 3,route a)。異構體3na’的形成(當R’=H時)可以用π-allyl-Pd III→III’的異構化來解釋,這可能是由于較低的位阻所致(Scheme 3,route b)。此外,氘標記實驗的結果支持了上述機理。

(圖片來源:Chem. Sci.)
李朝軍課題組利用簡單的腙作為碳負離子等價物,通過高選擇性的碳-碳σ鍵斷裂實現了鈀催化的亞甲基環丙烷的加氫烷基化。該方法反應條件溫和、具有良好的官能團相容性,可以高收率得到C-烷基化的末端烯烴。