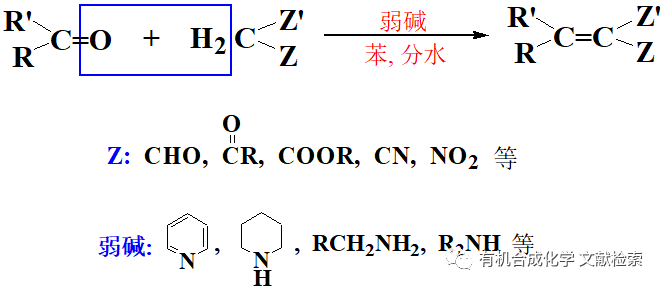
烯烴是有機合成中最重要的合成砌塊之一,尤其是末端烯烴因其易于獲得和廣泛的反應活性,在藥物合成、天然產物修飾以及高值分子構建中發揮著關鍵作用。然而,相較于內烯烴,末端烯烴的不對稱烯丙位C(sp3)–H氧化一直是一項懸而未解的挑戰。這類反應若能實現,不僅可直接轉化為光學純的烯丙醇或其衍生物,還能進一步通過雙鍵轉化拓展合成空間,具備重要的策略意義和實際價值。
早期如White 等人(2008年)提出了鈀-鉻雙金屬催化體系,但面臨ee值不高、底物過量、區域選擇性不足等瓶頸。Kharasch–Sosnovsky(K-S)反應盡管被廣泛研究,但其在末端烯烴中的應用仍因活性差、副反應嚴重、對映選擇性差而受限。此前已報道的銅/手性配體催化體系(如Sp-Box/Cu)即使優化至極限,也需要10倍烯烴才能獲得中等收率和選擇性,更難滿足實際應用所需。

圖片來源:JACS
近年來,通過光催化或仿生策略提升烯丙位C–H氧化反應的區域與對映選擇性成為新方向,但大多仍局限于內烯烴或苯乙烯類體系。由于末端烯烴的電子結構和立體環境不同于內烯烴,如何在不依賴過量底物的前提下,實現其高效、區域及對映選擇性良好的氧化反應,亟需新的催化體系突破這一“瓶頸”。
本研究發展了一種銅催化的高效且對映選擇性良好的末端烯烴烯丙位C(sp3)–H氧化反應體系。研究團隊構建了以CuCl為金屬源,手性Box類配體L1為配體,PivOOtBu為自由基前體,DIADH?為還原劑,并特別引入體積龐大的[B?Im(C?F?)?]?弱配位陰離子構建陽離子銅催化劑。這種設計的初衷是通過立體位阻效應抑制Cu(I)對自由基的猝滅,進而保證烯丙自由基向Cu(II)羧酸配位物的高效轉移,實現區域與對映控制。
優化實驗表明,在t-BuOH/PhCl溶劑中、16 °C下,該催化體系對3-苯基取代末端烯烴1a表現出優異的催化活性和選擇性,獲得目標產物3a的收率高達76%,對映選擇性為94% ee,且b/l(支鏈/線性)選擇性超過20:1。

圖片來源:JACS
隨后作者系統評估了催化劑結構、陰離子種類、還原劑、氧化劑及溶劑等參數的影響,進一步證實了B?Im(C?F?)?在提升催化效率與抑制副反應中的關鍵作用。在此基礎上,團隊拓展了底物適用范圍,包括各種苯環取代基、雜芳基以及復雜藥物骨架底物,并在多個實例中實現了93–99%的對映選擇性。

圖片來源:JACS
本項研究在多個層面上推動了不對稱C–H氧化反應的發展。首先,在合成策略上,該方法實現了在不使用底物過量的條件下對末端烯烴進行高效不對稱氧化,一舉解決了K-S反應體系需大量烯烴和選擇性差的問題,具有極強的現實應用潛力。其次,通過合理設計陽離子銅催化體系,揭示了陰離子大小和配位能力在催化循環中對自由基路徑選擇與催化效率的深遠影響,為今后催化劑設計提供了新的思路。
更值得一提的是,該方法不僅適用于常規芳基烯烴,更能擴展至雜芳基、天然產物、藥物分子等復雜底物,顯示出優越的底物普適性與實用性。例如其構建的手性烯丙酯骨架被成功用于合成GCS抑制劑PDMP及抗抑郁藥Reboxetine的重要中間體。此外,該研究還結合機制實驗與DFT理論計算,從自由基生成與捕獲、Cu(I)/Cu(II)還原循環等多個角度全面解析了催化過程中的關鍵步驟,為深入理解自由基不對稱氧化反應機制奠定了堅實基礎。

圖片來源:JACS
標題:Copper-Catalyzed Highly Efficient and Asymmetric Allylic C–H Oxidation of 3-Aryl-Substituted Terminal Alkenes
作者:Yibo Zhou, Pinhong Chen, and Guosheng Liu*
鏈接:https://doi.org/10.1021/jacs.5c05382