甲酰胺制備醛
有機金屬化合物(有機鎂、有機鋰化合物)可與過量的原甲酸脂反應,首先生成縮醛,繼而用硫酸水解成醛,廣泛用于脂醛及芳醛的合成,產率達55%-90%。
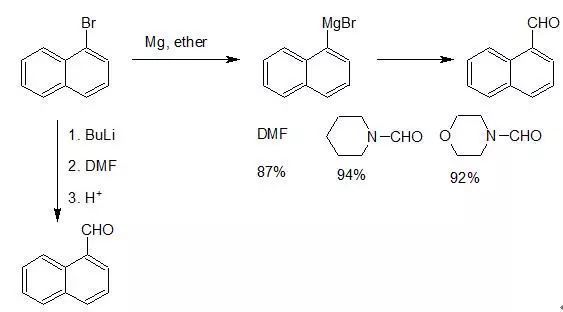
原甲酸脂外,甲酰胺、乙氧亞甲基苯胺(ethoxymethyleneaniline,C6H5N=CHOC2H5)等均為常用的甲酰化試劑。其中以甲酰胺的應用最為常見,常稱為Bouveault反應。芳鹵或乙烯鹵化物與丁基鋰發生金屬化反應,生成芳基鋰或乙烯基鋰,后者與二甲基甲酰胺反應,高產率地生成相應的醛。醛或酚醚的鄰位氫比較活潑,可與丁基鋰直接進行金屬化反應,繼而甲酰化和水解,是鄰羥基或鄰烷氧基苯甲醛的良好合成方法。
將鹵化物變為鎂(Grignard試劑)或鋰化物等,進行酰化可以合成醛酮。由有機金屬化合物合成醛的甲酰化試劑以甲酸酯類、甲酰胺類用得較普遍,以DMF或N-甲酰哌啶使用較為方便。常用的甲酰化試劑有:FCHO,(HCO)2O,HCOOCOCH3,CH(OCH3)3,HCO2C2H5,HCO2Li,PhN=CHOC2H5,Ph-N(CH3)-CHO,DMF,Py-N(CH3)-CHO,N-甲酰基哌啶,N-甲酰基嗎啉。
相關文獻
G. A. Olah, Synthesis, 1984, 228;
G. A. Olah, Angew. Chem., Int. Ed. Engl., 20,878,1981; Org. Synthesis., 64, 114, 1985;
M. Bogavac, Tetrahedron Lett., 1984, 1843
E. A. Evans, Chem. Ind., 1957, 1596
Bouveault反應
反應實例
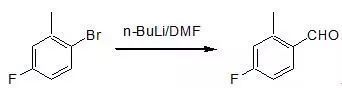
n-BuLi (2.6M in hexane, 57 mL, 143 mmol) was addedover 15 minutes to a THF solution (200 mL) of 2-methyl-4-fluorophenylbromide (24.5 g, 130 mmol), cooled to -78.deg. C. andallowed to stir 1 hour at -78 oC. DMF (26.61 gm, 364 mmol) was then added over 2 minutes, andthe solution was allowed to stir another hour. The reaction was quenched with NH4Cland warmed to room temperature. 10% HCl was added until the solution becameacidic. The mixture was diluted withether and the organic layer washed with water and brine, then dried over MgSO4,filtered and concentrated to give an orange oil. The oil was purified by distillation (bp=69 oC.at 7 mm Hg) to afford aldehyde 4-fluoro-2-methylbenzaldehydeas a clear liquid (13.3 g, 74%). TLC: Rf 0.30 (10% EtOAc in hexane)
weinreb酰胺制備酮
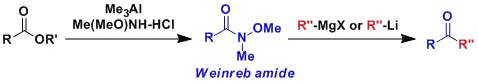
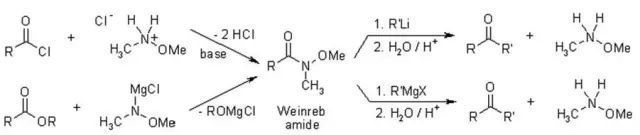
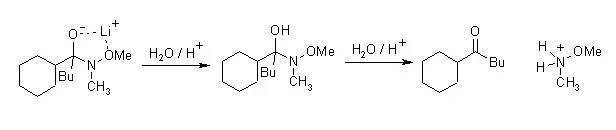
N-甲氧基-N-甲基酰胺俗稱Weinreb酰胺、它能與Grignard試劑或有機鋰試劑反應生成酮。酰鹵或是酯中加入兩倍當量的格式試劑或是有機鋰試劑的話會得到醇,而Weinreb酰胺則能夠避免這種過度的加成。
用氫化鋁鋰或Red-Al、DIBAL等還原Weinreb酰胺的話,會得到醛。
反應機理
Weinreb酰胺和有機金屬試劑的加成物由于甲氧基的存在形成一個五元環的螯合物相對穩定,使反應不再繼續。
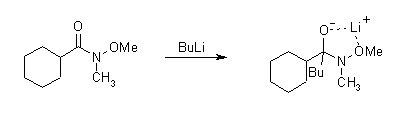
配合物水解則得到酮。
反應實例
一、由Weinreb酰胺還原合成醛反應
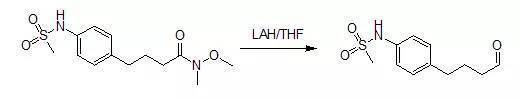
4-[4-(methanesulfonamido)phenyl]butyraldehyde
A mixture of 4.20 g (14 mmol) of4-[4-(methanesulfonamido)phenyl]butyric acid, N-methoxy-N-methylamide and 100 mLof anhydrous tetrahydrofuran was stirred under nitrogen with cooling in an icebath as 17.5 mL (17.5 mmol) of 1Mlithium aluminum hydride in tetrahydrofuran was added gradually by syringe. After 0.75 hours, 70 mL of 5percent potassiumhydrogen sulfate solution (aqueous) was added cautiously by syringe. Themixture was then removed from the ice bath, diluted with 150 ML of water, andshaken with 150 mL of ethyl acetate. The milky aqueous phase was extracted withan additional 50 mL of ethyl acetate. The combined organic fractions werewashed successively with 2*100 mL of 1N hydrochloric acid, then 50 ML ofsaturated aqueous sodium bicarbonate solution, and finally 50 ML of saturatedaqueous sodium chloride solution. The organic phase was dried over magnesiumsulfate, filtered, and concentrated in vacuo. Flash chromatography of theresidue on silica gel (elution with 3:2 hexane-EtOAc) yielded 2.47 g (73%) of an oil; homogeneous by TLC in1:1 hexane-EtOAc).Upon storage in the freezer, solidification occurred (mp 41~44oC.).
【US5756507】
二、由Weinreb酰胺還原合成酮
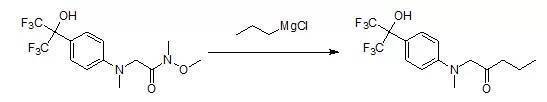
1-(Methyl{4-[2,2,2-trifluoro-1-hydroxy-1-(trifluoromethyl)ethyl]phenyl}amino)-pentan-2-one
To a solution of [N-1--methoxy-N-1-N-2-dimethyl-N-2-- {4- [2,] 2,2- trifluoro-1-hydroxy-1-(trifluoromethyl)ethyl]phenyl}glycinamide (240 mg, 0.64 mmol) in CH2Cl2(3.5 mL) at 0 oC under a nitrogen atmosphere was slowly added asolution of propylmagnesium chloride (1.6 mL, 2M in Et2O). After stirring 3 hours, the reaction mixturewas poured into 1 M HCI andextracted twice with EtOAc. The organic layer was washed with brine, dried (NA2SO4),filtered, and concentrated. Purification by flash chromatography (2: 1 EtOAc/hexanes) afforded the title compound as a dark yellow waxy solid (110 mg,48%).
【WO2003/99769】
三、
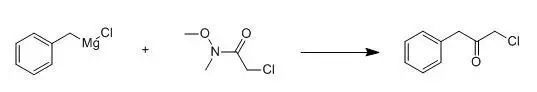
Benzylmagnesium chloride (1.0 M in diethyl ether, 50.0 mL) was treated with 2-chloro-N-methoxy-N-methylacetamide (5.16 g, 37.5 mmol) in THF (200 mL) at -78℃drop wise. The reaction was allowed towarm slowly to room temperature overnight and quenched with 1N hydrochloric acid. The layers wereseparated and the organic phase dried (Na2SO4), filtered,and the filtrate was concentrated under reduced pressure. The residue was purified by flash columnchromatography (silica gel, elution with hexanes) to provide the title compound.
四、
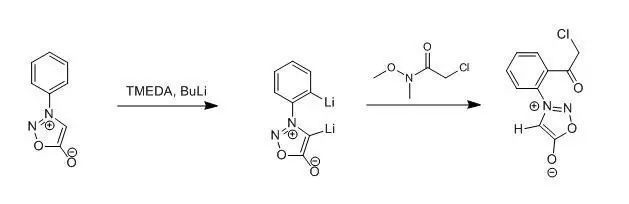
To a stirred solutionof 3-phenylsydnone (1) (0.25 g,1.54 mmol) in dry THF (100 ml) at -78℃ under an atmosphere of dry nitrogengas was added N, N, N’, N’-tetramethylethylenediamine (0.29 ml, 1.93 mmol) then n-butyllithium (2.31 ml, 3.47 mmol, 15 Min pentane) dropwise. After 0.5 h, theappropriate 2-chloro-N-methoxy-N-methylacetamide (1.93 mmol) was added to thegolden yellow solution and, after a additional 1h, the mixture was quenchedwith aqueous hydrochloric acid (100ml, 10% v/v) then extracted withdichloromethane (3*100 ml). the combinedorganic layers were dried (MgSO4) and the solvent removed in vacuo to affordthe corresponding o-acylated sydnone 4 as an oil which was puriied by columchromatography to afford colorless crystals. Yield 86%.
【Turnbull,Kenneth; Sun, Congcong; Krein, Douglas M.; Tetrahedron Lett., EN; 39; 12; 1998;1509-1512】
五、通過Weinreb酰胺的還原得到醛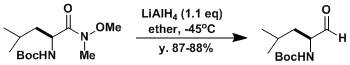
裝備有溫度計、攪拌子、滴液漏斗、空氣冷凝回流管的5L圓底瓶中,通入氬氣保護氣后,加入LiAlH4(0.44 mol)以及無水乙醚(1.5L)。室溫下攪拌一小時后,冷卻到-45℃。將Boc-亮氨酸Weinreb酰胺(~100g, 約0.4mol)的無水乙醚溶液(300mL)、保持反應溫度在-35℃以下緩慢滴加。加完后移去冷卻槽,攪拌下讓溫度緩慢回復到5℃。再一次冷卻降溫到-35℃,將KHSO4(96.4g, 0.71mol)的水溶液(265mL)緩慢滴加,這時保持溫度不要超過零度。去掉冷卻槽,繼續攪拌一小時。用硅藻土過濾反應液,用五百毫升乙醚洗凈固體殘渣。得到的有機溶液用1N鹽酸(350mL)在5℃下洗凈三次,再用飽和小蘇打溶液(350mL)和飽和食鹽水(350mL)先后分別洗凈三次,用無水硫酸鎂干燥除水。蒸干溶劑后,得到油狀的Boc-L-Leucinal(69-70g,產率87-88%)。生成物放在冰柜(-17℃)保存。
【 Goel, O. P.; Krolls, U.; Stier, M.; Kesten, S. Org. Synth. 1989, 67, 69.】
參考文獻
一、化學空間:https://cn.chem-station.com/reactions/%e8%bf%98%e5%8e%9f%e5%8f%8d%e5%ba%94/2014/06/%e6%b8%a9%e5%8b%92%e4%bc%af%e9%85%ae%e5%90%88%e6%88%90-weinreb-ketone-synthesis.html
二、藥明寶典