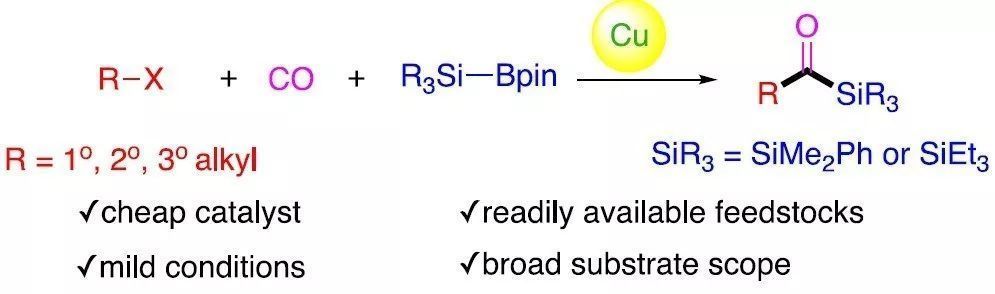
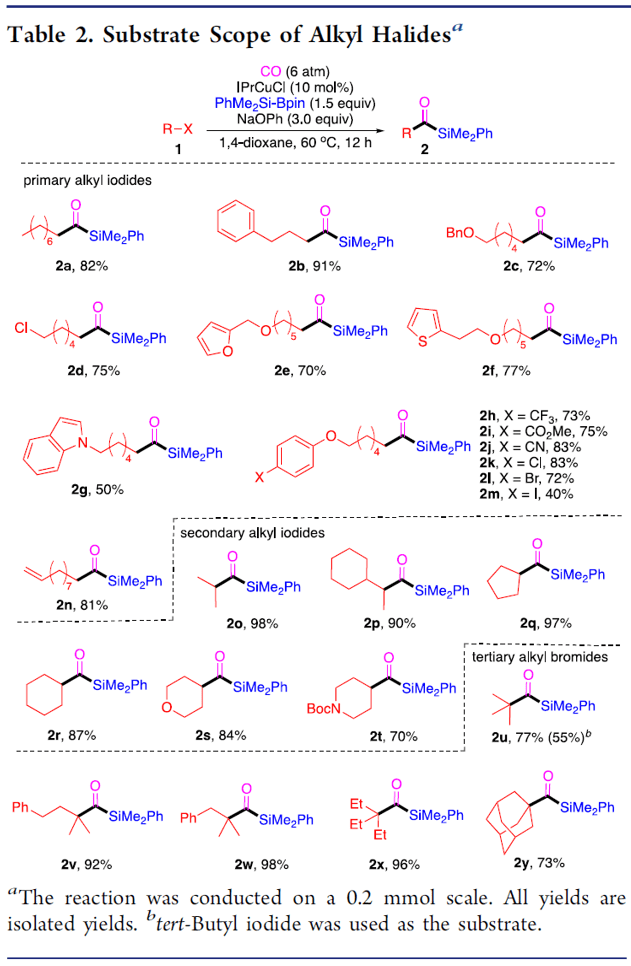
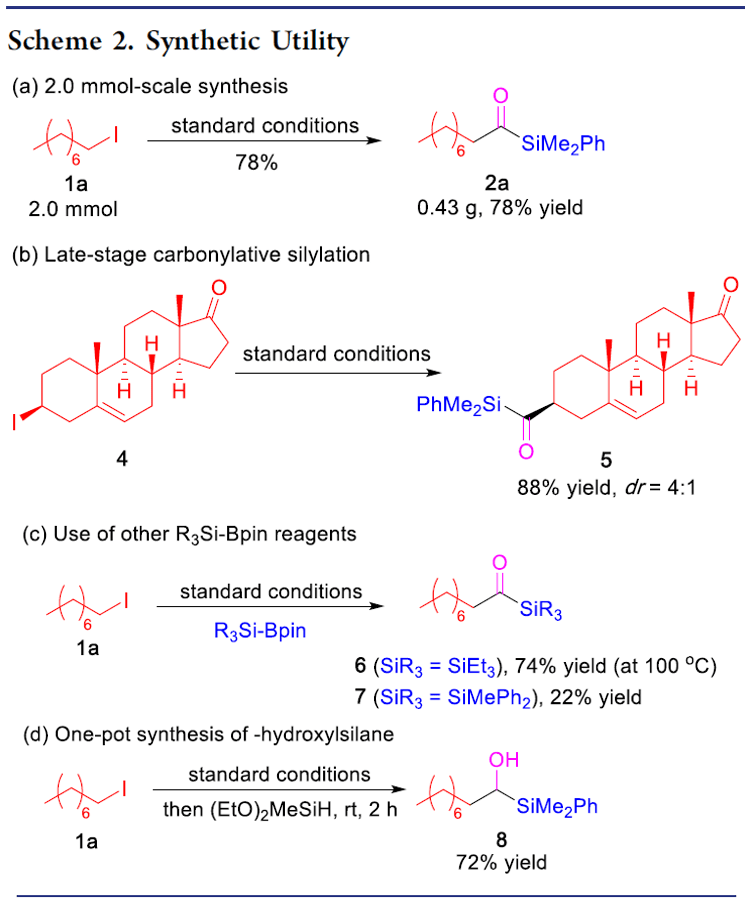
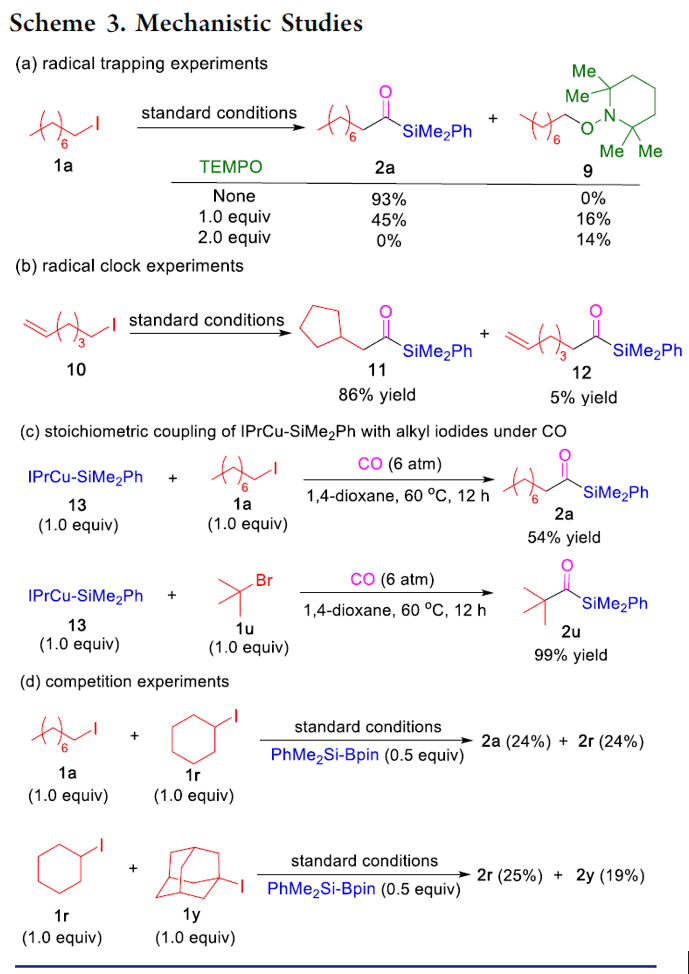
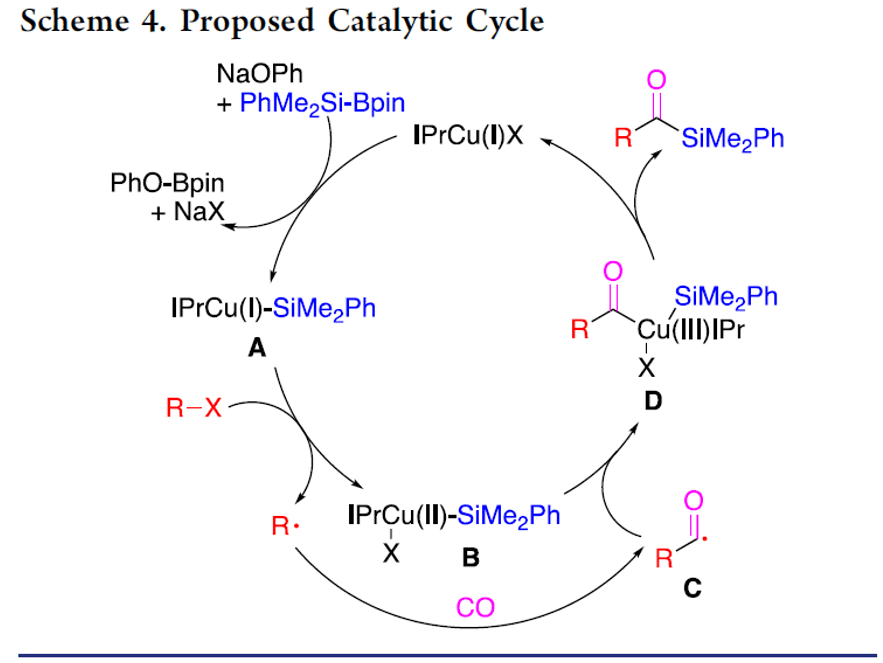
銅催化鹵代烷基的羰基硅烷化反應(yīng)
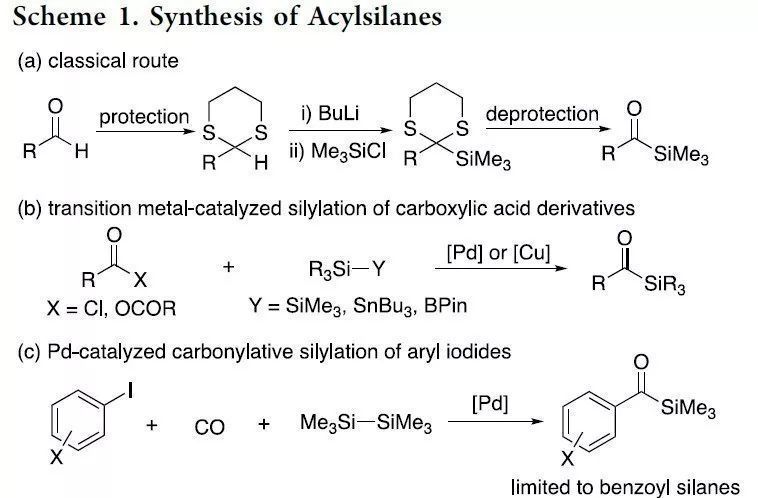
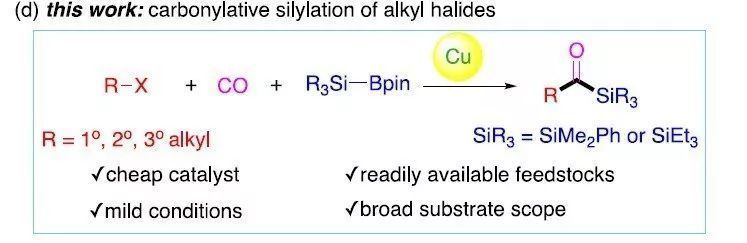
本文作者:杉杉 導(dǎo)讀 近日,伊利諾伊大學(xué)Mankad Neal教授課題組在JACS發(fā)表論文,報道了Cu催化下,實現(xiàn)未活化烷基鹵化物的羰基硅烷化反應(yīng),高效地合成了烷基取代的酰基硅烷衍生物。在溫和的反應(yīng)條件下可以耐受多種官能團(tuán),伯、仲和叔烷基鹵化物均可適用。此外,該方法不僅合成不同甲硅烷基的酰基硅烷衍生物,同時可將產(chǎn)物還原為相應(yīng)的α-羥基硅烷衍生物。機理研究表明,甲硅烷基銅中間體通過單電子轉(zhuǎn)移活化烷基鹵,形成烷基自由基中間體,進(jìn)而進(jìn)行羰基硅烷化反應(yīng)。 "Cu-Catalyzed Carbonylative Silylation of Alkyl Halides: Efficient Access to Acylsilanes Li-Jie Cheng, Neal P. Mankad* J. Am. Chem. Soc. 2020, 142, 1, 80-84. DOI:10.1021/jacs.9b12043" 正文 有機硅烷化合物廣泛應(yīng)用于有機合成和材料科學(xué)中,含有酰基硅烷的結(jié)構(gòu)單元作為典型代表,Brook重排是制備此類骨架的常用方法。然而,大多數(shù)已報道文獻(xiàn)中存在一些不足,如需多步制備、底物范圍窄等。Brook和Corey報道的經(jīng)典合成路線中,從醛類底物中合成酰基硅烷衍生物,但需要繁瑣的保護(hù)/脫保護(hù)過程(Scheme 1a)。盡管可將含有陰離子甲硅烷親核試劑直接加成到羧酸衍生物中(一步合成酰基硅烷衍生物),但通常需要化學(xué)計量的甲硅烷基銅試劑或甲硅烷基鋰試劑(這類試劑會限制官能團(tuán)的兼容性)。已報道文獻(xiàn)中,鈀催化下,可實現(xiàn)酰氯與乙硅烷或甲硅烷基錫試劑的甲硅烷基化反應(yīng),合成酰基硅烷衍生物。此外,Riant通過使用甲硅烷基硼烷試劑,在銅催化下,實現(xiàn)酸酐甲硅烷基化反應(yīng),獲得苯甲酰基硅烷衍生物(Scheme 1b)。盡管這些方法可以在溫和條件下簡單地合成酰基硅烷衍生物,但由于底物對水分敏感(如酰氯或酸酐),從而限制了它們的應(yīng)用范圍。為了克服這類問題,羰基化制備酰基硅烷作為一種高效的方法(可將羰基直接引入簡單的前體中)。早期,Seyferth和Weinstein報道了通過有機鋰化合物在-110℃時進(jìn)行羰基化反應(yīng),從而獲得酰基硅烷衍生物。此后,Beller報道了更為實用的鈀催化體系,通過芳基碘化物的羰基硅烷化反應(yīng),獲得苯甲酰基硅烷(Scheme 1c)。但是,烷基鹵化物底物尚未研究,可能是由于烷基親電試劑在羰基化反應(yīng)中,氧化加成速度非常慢,并且存在β-消除的競爭關(guān)系。 基于前期報道工作總結(jié)(銅催化炔烴與鹵代烷的羰基化C-C偶聯(lián)反應(yīng)和未活化烷基鹵的還原羰基化反應(yīng)),作者發(fā)現(xiàn)在此類反應(yīng)中,通常由烷基鹵生成酰基后再與各種有機銅親核試劑偶聯(lián),得到相應(yīng)的羰基類化合物。作者假設(shè),通過銅催化劑和甲硅烷基硼烷反應(yīng)生成親核甲硅烷基銅,是否可與鹵代烷反應(yīng)制備酰基硅烷衍生物(Scheme 1d)。在本文中,作者通過銅催化的羰基硅烷化過程,從烷基鹵化物直接催化合成烷基取代的酰基硅烷衍生物。 首先,作者以1-碘辛烷1a與PhMe2Si-Bpin作為模型底物,在含有CO(6 atm)下反應(yīng),經(jīng)過大量的條件篩選發(fā)現(xiàn)(Table 1),當(dāng)使用IPrCuCl作為催化劑,以NaOPh作為堿,以二氧六環(huán)作為溶劑時,可獲得93%收率的酰基硅烷2a(entry 1)。當(dāng)不加入銅催化劑時,則不發(fā)生反應(yīng)(entry 2)。而對NHC配體篩選中(entries 3-6),使用ClIPr配體時收率略低,而使用SIPr和MeIPr配體時收率大幅下降。有趣的是,使用空間位阻較小的IMes配體時,沒有目標(biāo)產(chǎn)物,但獲得較高收率烷基硅烷3a。如果將底物1a的碘取代改為溴或氯時,導(dǎo)致更差的反應(yīng)結(jié)果(entries 7-8)。而對堿的篩選中(entries 9-11),NaOMe、NaOtBu、LiOMe均沒有獲得較好的結(jié)果。而將PhMe2Si-Bpin的量減少至1.2倍當(dāng)量或?qū)aOPh降至2.0倍當(dāng)量時,產(chǎn)率都略有降低(entries 12-13)。而對溶劑的篩選中(entries 14-15),二氧六環(huán)作為最佳溶劑。如果將反應(yīng)在室溫下進(jìn)行,則導(dǎo)致大量烷基硅烷副產(chǎn)物(entry 16)。值得注意的是,如果將反應(yīng)在3 atm或1 atm大氣CO壓力下也可獲得所需產(chǎn)物,但收率略有降低(entries 17-18)。 在獲得上述最佳反應(yīng)條件后,作者開始對底物進(jìn)行了擴展(Table 2)。反應(yīng)結(jié)果表明,烷基碘化物末端含有苯基(2b)、芐基醚(2c)、氯烷基(2d)、末端烯烴(2n)等,均獲得較好的結(jié)果。而含有雜環(huán)的底物也能夠與體系兼容,如呋喃(2e)、噻吩(2f)、吲哚基(2g)等。末端苯基(2h–2l)含有三氟甲基、酯基、氰基、氯基、溴基取代時,也可獲相應(yīng)的目標(biāo)產(chǎn)物,但碘取代的底物(2m)存在一定的副反應(yīng)。隨后,作者將伯烷基碘擴大到仲烷基碘,底物仲烷基碘(2o–2r)不受位阻影響,同時環(huán)烷基碘(2s、2t)也與體系兼容。隨后,叔烷基親電體(2u–2y)也獲得了令人滿意的結(jié)果,同時具有更高的收率。 為了驗證該方法的適用性,作者進(jìn)行了如下實驗(Scheme 2)。首先,克級實驗同樣以高收率獲得了預(yù)期的酰基硅烷產(chǎn)物2a(Scheme 2a)。此外,該方案可對天然產(chǎn)物或藥物進(jìn)行后期的修飾。如雌酮衍生物4,在上述標(biāo)準(zhǔn)條件下,以優(yōu)異的產(chǎn)率獲得酰基硅烷5(Scheme 2b)。此外,除了PhMe2Si-Bpin作為甲硅烷基來源外,活性較低的Et3Si-Bpin(高溫下)也可作為偶聯(lián)底物(Scheme 2c)。如果在反應(yīng)完成之后,向反應(yīng)混合物中加入氫硅烷,可進(jìn)一步還原酰基硅烷(Scheme 2d)。因此,該方案也作為合成α-羥基硅烷(烷基鹵化物作為底物)的有效方法。 為了進(jìn)一步驗證機理的正確性,作者進(jìn)行了相關(guān)的對照實驗(Scheme 3)。當(dāng)將TEMPO加入反應(yīng)中時,未獲得目標(biāo)產(chǎn)物,并且通過1H NMR檢測發(fā)現(xiàn)自由基中間體9的存在(Scheme 3a),從而證明了自由基機理。同時,使用碘代烷10進(jìn)行自由基反應(yīng),獲得86%收率的環(huán)化產(chǎn)物11(Scheme 3b)。此外,通過相關(guān)實驗制備了甲硅烷基銅物種IPrCu-SiMe2Ph中間體13,并在CO與伯烷基碘(1a)和叔烷基碘(1u)進(jìn)行相關(guān)反應(yīng),分別獲得54%和99%收率的酰基硅烷(Scheme 3c),表明甲硅烷基銅中間體可促進(jìn)伯和叔烷基碘的單電子轉(zhuǎn)移過程,進(jìn)而進(jìn)行自由基羰基化反應(yīng)。此外,由于伯,仲和叔烷基碘之間存在的競爭關(guān)系,因此作者對反應(yīng)的相對速率也進(jìn)行了研究,但反應(yīng)結(jié)果獲得相似的收率產(chǎn)物(Scheme 3d)。 根據(jù)上述的實驗和相關(guān)文獻(xiàn)的查閱,作者提出了一種可能的反應(yīng)機理(Scheme 4)。首先,通過PhMe2Si-Bpin與IPrCuOPh(由IPrCuCl和NaOPh形成)反應(yīng)生成甲硅烷基銅(I)配合物A。隨后,配合物A和烷基鹵化物之間的進(jìn)行單電子轉(zhuǎn)移(SET),形成甲硅烷基銅(II)配合物B和一個烷基自由基R·。緊接著,自由基R·進(jìn)行羰基化反應(yīng),得到酰基自由基C,再與銅(II)配合物B配位,形成銅(III)中間體D。最后,經(jīng)還原消除得到酰基硅烷,同時再生銅(I)催化劑,從而實現(xiàn)催化循環(huán)。 總結(jié) Mankad教授課題組報道了通過Cu催化下的羰基硅烷化反應(yīng),從未活化的烷基碘底物合成烷基取代的酰基硅烷衍生物。該方法不僅適用于伯、仲和叔烷基鹵底物,同時還具有良好的官能團(tuán)兼容性。此外,通過雌酮衍生物的后期羰基硅烷化反應(yīng)以及將產(chǎn)物還原為相應(yīng)的α-羥基硅烷衍生物,進(jìn)一步證明了該方法的綜合實用性。機理研究表明,甲硅烷基銅中間體通過單電子轉(zhuǎn)移活化烷基鹵,形成烷基自由基中間體,進(jìn)而進(jìn)行羰基硅烷化反應(yīng)。