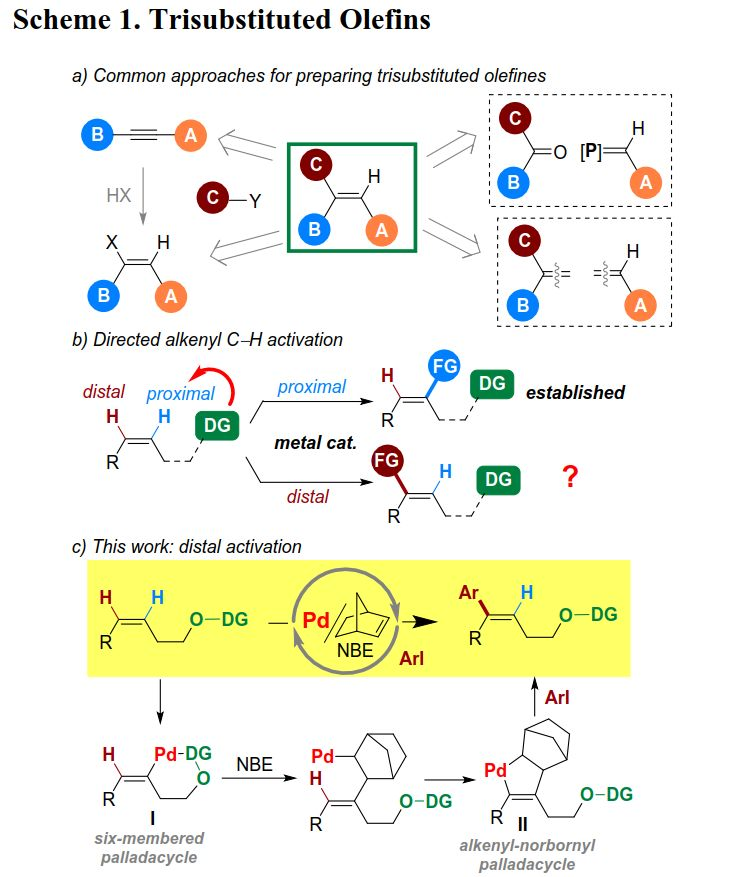
三取代烯烴是許多天然產物以及生物活性化合物的常見結構片段,也常作為合成中間體構建其他官能團,如環氧化物、(氮雜)環丙烷等。目前,三取代烯烴主要通過羰基的烯化、炔烴的加氫官能團化、交叉偶聯和烯烴復分解等方法來制備(Scheme 1a)。在不存在空間位阻或電性偏向的情況下,區域和立體選擇性合成三取代烯烴尤為重要。由于鄰近效應,通過導向基團可以提高烯基C-H活化的反應性和位點選擇性(Scheme 1b)。迄今為止,許多過渡金屬絡合物(如Ru、Pd、Rh、Ir、Fe和Co)可以實現近端烯基C-H鍵的官能團化。然而,對于通過遠端烯基C-H鍵的位點選擇性活化實現區域選擇性合成三取代烯烴的研究鮮有報道。近日,美國芝加哥大學董廣彬課題組通過鈀/降冰片烯(Pd/NBE)協同催化實現了遠端烯基C-H官能團化(Scheme 1c),該成果發表于近期J. Am. Chem. Soc.(DOI: 10.1021/jacs.9b11479)。
(圖片來源:J. Am. Chem. Soc.)
最近,由Catellani課題組開發的Pd/NBE催化劑已廣泛用于芳烴的官能團化。2015年,余金權課題組和作者分別報道了Pd/NBE催化下由定向鄰位鈀化引發的間位選擇性C-H活化方法。然而,這種遠端的C-H官能團化策略是否可以擴展到烯烴等芳香族化合物之外的底物?與芳烴相比,烯烴C-H鍵的活化可能存在諸多挑戰:首先,與氧化劑或親電試劑反應時,反應性更高的烯烴C=C鍵易發生各種π斷裂轉化;其次,烯烴的π鍵是比芳烴更好的鈀配體,易導致鈀介導的副反應;此外,NBE插入后,烷基-Pd中間體易通過遷移插入與烯烴形成環丙烷,而非與遠端的C-H鍵發生鈀化。為了應對上述挑戰,需要適當的導向基團(DG)實現快速且可逆的近端C-H鈀化。此外,作為反應性配體,NBE的結構對減少副反應很重要。首先,作者以順式烯烴1a作為初始底物探索了遠端烯烴C-H官能團化(Table 1)。通過條件優化作者發現,當以Pd(OAc)2/N4為催化劑、3-CF3-2-吡啶酮為添加劑、AgOAc作為鹵化物清除劑時,反應可以76%的收率得到C-H芳基化產物3a。盡管醛衍生的DG2-4的反應性很低,但環己酮衍生的DG5卻顯示較好的結果。對照實驗表明,鈀和NBE對反應必不可少,并且在不加吡啶酮時收率顯著降低。不存在NBE時,反應主要形成Heck產物(Z)-3a;而標準條件下,Heck反應被顯著抑制。

(圖片來源:J. Am. Chem. Soc.)
在確定最佳反應條件后,作者研究了烯烴的適用范圍(Table 2)。含缺/富電子取代基的苯乙烯基烯烴、六元或八元環烯以及順式1,2-二烷基取代的烯烴是適合的底物。盡管芳烴上有潛在的副反應,但苯基取代的底物仍可得到遠端烯烴芳基化產物。除脂肪族醇外,芐醇衍生的底物也反應良好;除仲醇外,伯醇衍生的底物也能參與反應,而與伯醇衍生的鏈烯的反應性較低。此外,當用改良的DG后,烯丙基和高烯丙基保護的胺也反應良好。
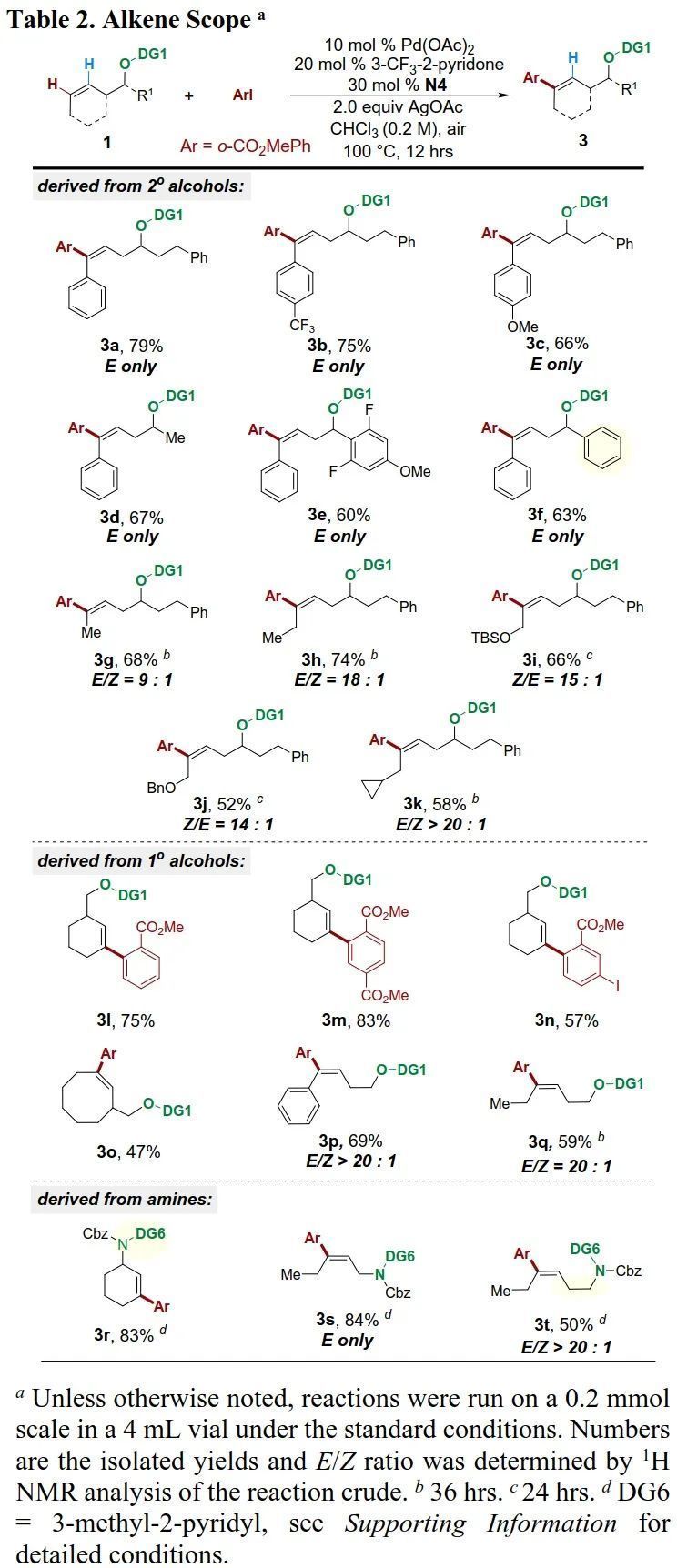
(圖片來源:J. Am. Chem. Soc.)
接下來,作者探討了芳基碘化物的適用范圍(Table 3)。含鄰位吸電子基團、缺電子的芳基碘化物均可以良好的收率和優異的E/Z選擇性得到遠端芳基化產物,而富電子的芳基碘化物則收率較低且選擇性中等。除酯外,鄰位基團還可以是硝基、酮、叔酰胺和Weinreb酰胺等。該反應也可以耐受雜芳烴,如噻吩和吡啶以及碘、溴和氯取代基。在標準條件下,不含鄰位吸電子基團的芳基碘化物的反應性較低。除芳基化外,在稍加改良的條件下,還可以實現碘甲烷和溴乙酸甲酯的遠端C-H烷基化。
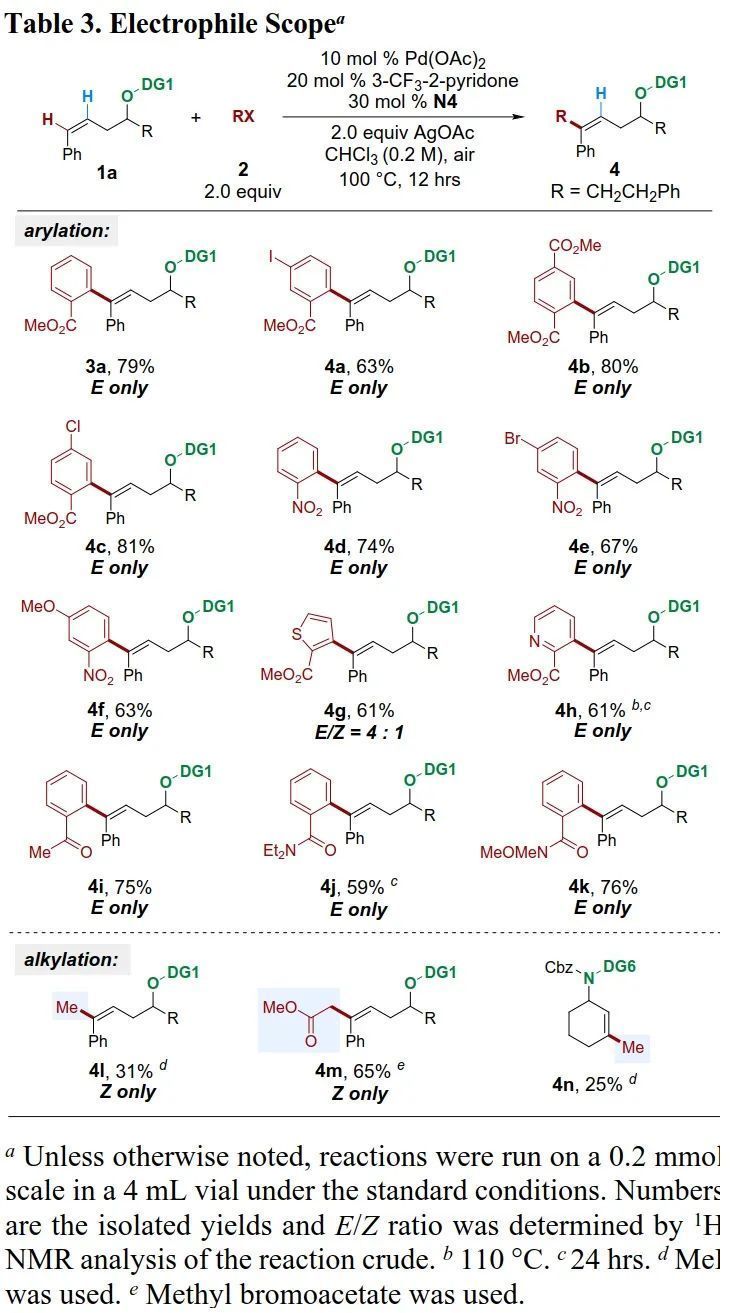
(圖片來源:J. Am. Chem. Soc.)
該反應的主要競爭性反應途徑是定向的Heck反應(Scheme 2a)。將芳基-Pd(II)定向遷移至烯烴后經.