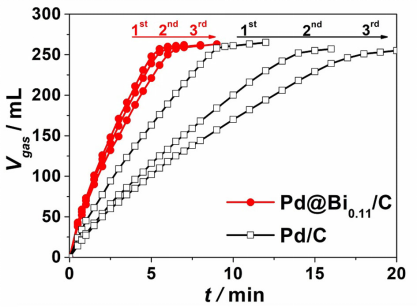
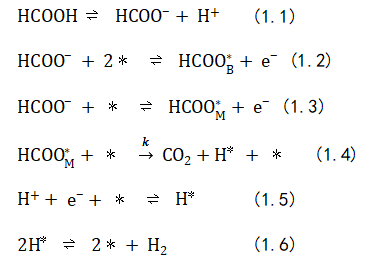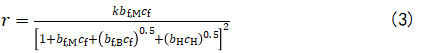論文DOI:10.1021/acscatal.0c00225 本文從分析甲酸脫氫-產氫的反應機制出發,提出甲酸脫氫-制氫鈀基催化劑的設計指導原則,即催化劑對甲酸根HCOO-的吸附能力應適當增強,對H或CO的吸附能力應適當削弱。根據這一設計原則,將Bi,Sb和Pb等主族半金屬元素修飾在Pd的表面被選定為理想催化劑。本文以Bi修飾為例,利用不可逆吸附,在自制的Pd/C催化劑Pd表面修飾Bi原子,獲得了具有優異性能的甲酸制氫Pd@Bi/C催化劑。理論計算與原位表面增強紅外光譜表明了設計原則在實際合成的Pd@Bi/C催化劑上得到了集中體現。該工作建立了催化劑結構與性能之間的構效關系,深化了對甲酸制氫反應機制的認識。新能源的開發與利用成為實現可持續發展的技術保障。其中,氫能是高效、經濟、清潔的新能源,受到了科學家和工業界的廣泛關注。然而,將氫能直接存儲在氫氣中具有重大安全隱患,給實際生產、運輸和使用帶來極大不便。在適當的催化劑幫助下,甲酸可以在常溫下分解,釋放出氫氣,因此甲酸被認為是一種比較高效、安全可控的儲氫材料。目前,最有效的甲酸制氫多相催化劑是鈀以及各種鈀基二元合金催化劑。在之前的研究中,本課題組首次報道了B摻雜的Pd催化劑是一種優秀的甲酸制氫催化劑(J. Am. Chem. Soc., 2014, 136, 4861.),后續的工作發現它也可促進二氧化碳電還原合成甲酸(J. Am. Chem. Soc., 2018, 140, 2880.),實現了同種催化劑對甲酸-二氧化碳的高效儲氫-釋氫的碳循環。然而,現有甲酸制氫催化劑的設計工作出現理論和實驗的斷裂:實驗工作以材料嘗試合成為中心,難以明確催化劑構效關系,無法有效指導后續工作;理論工作以模擬計算為中心,模型結構難以被實驗復現。因此,基于反應機理分析,提出一種經實驗驗證、可以被催化工作者理解和接納的催化劑設計原則對這一領域的發展具有重要意義。1.從甲酸分解制氫的反應機制出發分析了相關催化劑的設計原則,指出理想的甲酸制氫鈀基催化劑應具備以下特點:相較于純鈀,新型催化劑表面對甲酸根具有更強的的吸附能力,而對CO或H的吸附能力更弱。2.Bi、Pb或Sb等部分帶正電荷的M原子不可逆吸附在Pd納米粒子表面上形成的Pd@M/C催化劑,均可滿足設計原則, 也被DFT模擬計算結果初步驗證。實際制氫活性和穩定性顯著提升,體現了指導原則的普適性和催化劑合成的便利性。3.應用原位ATR-IR光譜跟蹤檢測了甲酸脫氫產氫過程的微量副產物CO在催化劑表面的累積,證實了相對于未修飾的Pd,Bi修飾后的Pd上CO的生成和累積得到了抑制。甲酸自分解反應在常溫下就可以在Pd的表面發生,由兩個平行反應組成,其一是生成CO2和目標產物H2的脫氫反應路徑,其二是生成催化劑毒物CO和H2O的脫水反應路徑。基于受到廣泛認可的電化學工作,Pd上甲酸脫氫反應路徑總結如下:2) 單重位吸附態甲酸根以及相鄰的裸露Pd位是C-H鍵解離反應的反應物;3) 橋式位吸附的甲酸根存在于催化劑表面,不直接參與反應,但是參與吸脫附平衡,可以轉化為單重位吸附態甲酸根;4) 考慮到反應發生的穩定電勢接近0 V vs.RHE,Pd的表面存在著大量吸附態H,占據著反應位點。由式1.4寫出反應速率方程,應用簡化的Langmuir吸附等溫式將(2)式展開,得式中和分別表示溶液中甲酸根和氫氣的濃度,,和分別是, 和H*的吸附常數。當甲酸根濃度不太高時,上式可以簡化
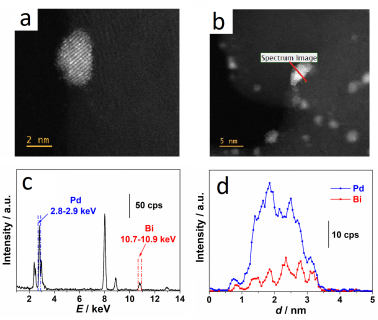
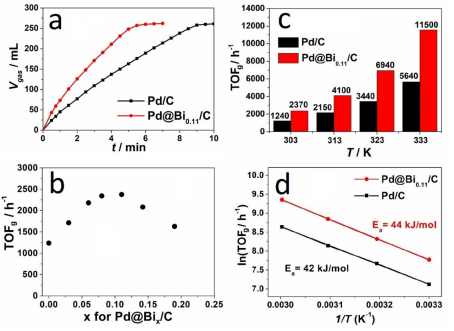
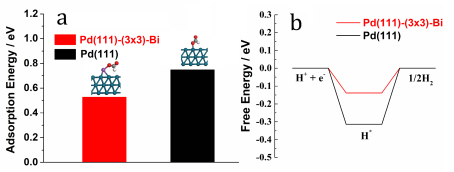
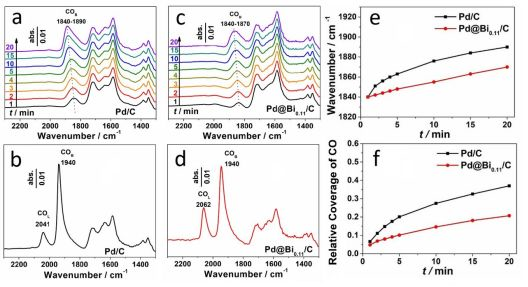
不難看出甲酸脫氫反應速率與甲酸根的吸附強度正相關,與H的吸附強度負相關。因此,需要增強Pd對甲酸根的吸附,減弱對H的吸附。另一方面,脫水反應路徑會生成強吸附物種CO,毒化催化劑表面,因此需要減弱催化劑對CO的吸附。已發表的理論工作表明,金屬材料對CO和H的吸附強度是正相關的。因此,設計原則仍然不失其簡單性。至此,我們得到甲酸制氫催化劑的設計指導原則:理想的甲酸制氫鈀基催化劑,相較于鈀,應具有更強的對甲酸根的吸附能力,更弱的對CO或H的吸附能力。查閱已經發表的電化學實驗和理論工作,Bi、Pb、Sb這一系列主族半金屬元素被選定為目標元素與Pd形成二元金屬催化劑。這一類元素對H、CO的吸附強度很弱,但是比Pd更親氧,更容易與甲酸根發生氧端吸附。另一方面,這一類元素與Pd接觸形成二元金屬體系后,會向Pd轉移電子,使得Pd費米能級附近的電子密度降低,進一步減弱Pd對CO、H物種的吸附強度。方程2表明存在一個最優的吸附原子覆蓋度,使產氫速率最大;而方程3表明在固定甲酸濃度下,存在最優的甲酸根濃度,使產氫速率最大。本工作將以Bi修飾的Pd作為模型催化劑說明催化劑設計原則的有效性。無需額外添加還原劑,將合成好的約2.3-nm Pd/C催化劑浸泡在Bi(NO3)3/HNO3溶液中,Bi就會隨機修飾在Pd顆粒的表面,這種合成方法十分成熟,最早可追溯到上世紀80年代相關的Pt上不可逆吸附Bi等原子。在Bi修飾之后,Pd金屬顆粒基本保持了2.5 nm的粒徑,未發生明顯團聚(Figure 1a,1b)。非原位的XPS測試表明,在空氣中,金屬顆粒表面的金屬原子都會被氧氣氧化。Bi幾乎完全以氧化態存在,說明Bi主要存在于可以直接與空氣接觸的金屬顆粒表層(Figure 1c,1d)。原位XAFS表明金屬顆粒表面的Pd在甲酸脫氫反應過程中會被還原,但無論在制氫條件下還是空氣中,Pd-Pd鍵的鍵長均與Pd單質相同,表明Bi并沒有破壞Pd的fcc晶體結構,沒有形成Pd-Bi合金結構(Figure 1e,1f)。這種Bi隨機點綴在Pd顆粒表面的結構可以直接在HAADF-STEM照片和EDS圖譜中看出(Figure 2)。圖1.Pd/C和Pd@Bi0.11/C催化劑的形貌結構和電子特性表征圖2.Pd@Bi0.11/C的高分辨STEM和能譜表征實驗表明,Bi修飾后的Pd/C催化劑對甲酸制氫反應表現出更高的催化活性(Figure 3a),催化活性隨Bi的覆蓋度存在極大值,極大值在Pd:Bi總原子比為9:1時取到(Figure 3b)。在優化的甲酸-甲酸根溶液中,303 K下Pd@Bi0.11/C上產氫的TOF值可達4350 h--1。值得注意,Bi對催化活性的提升效應隨溫度變化不大,修飾前后催化劑的表觀活化能幾乎不變(Figure 3c, 3d),這提醒我們表觀活化能往往無法反映催化劑的本征活性。甲酸制氫循環性測試表明Bi修飾后的催化劑具有更高的反應穩定性,這很可能與其減慢了CO在催化劑表面的累積速度有關(Figure 4)。圖4.Pd@Bi0.11/C和Pd/C催化劑甲酸制氫循環性能對照DFT計算結果表明,在Bi修飾在Pd表面后,甲酸根傾向于吸附在略帶正電的Bi原子上,與純Pd相比吸附能增強。值得注意,兩種表面上,甲酸根的吸附能都很弱(能量越負,吸附越強),這與實驗事實以及和我們上述推導的假設相符(Figure 5a)。另一方面,在Bi修飾在Pd表面后,H對金屬表面的結合能減弱(Figure 5b)。總之,DFT計算表明Bi修飾后,催化劑對甲酸根的吸附能力變強,對H的吸附能力變弱。另一方面,我們直接應用原位ATR-IR技術監測CO在反應過程中在催化劑表面的累積速率(Figure 6)。結果表明,Bi修飾后,CO在催化劑上的累積速率顯著下降,表明催化劑獲得了更高的反應選擇性和抗CO中毒能力。圖5. DFT計算得到的模型表面HCOOM*吸附能和H* 結合能圖。圖6.甲酸制氫反應過程中原位ATR-IR光譜跟蹤催化劑表面CO累積上述工作基于甲酸脫氫-產氫機制的動力學分析,提出了Pd基-二元催化劑的設計指導原則,即表面對甲酸根吸附增強但對H、CO吸附減弱的催化劑可促進甲酸分解產氫。據此,在自制Pd/C的Pd表面自發沉積Bi原子,獲得了具有優異性能的Pd@Bi/C催化劑。通過各種物理表征驗證其結構,通過DFT計算和原位紅外光譜表征進一步驗證上述指導原則的合理性。值得一提的是,該工作從反應機理、反應動力學分析的角度出發,為快速設計、篩選本證活性和穩定性同步提升的甲酸制氫催化劑提供了一種新思路。上述催化劑設計指導原則具有拓展性,不僅適用于鈀表面修飾異種金屬原子,也可適用于其它Pd基二元合金催化劑的理性合成。