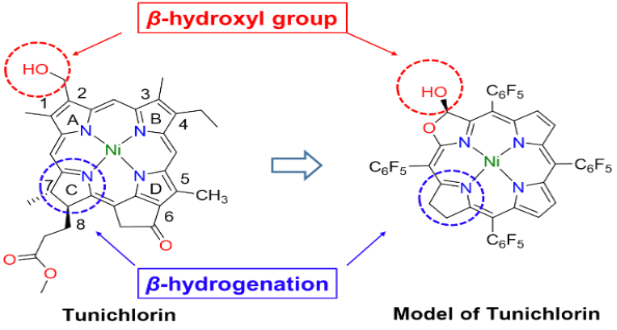
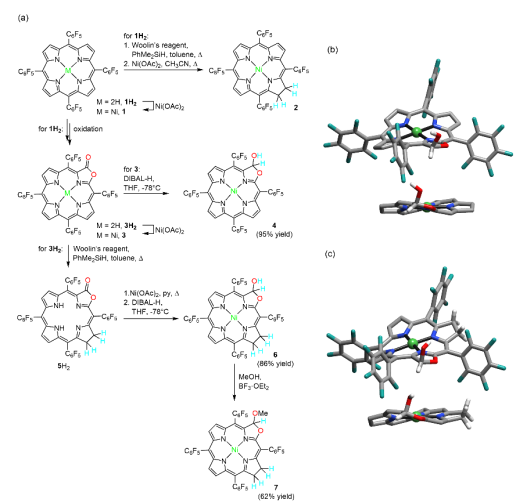
著名物理學家 Richard Feynman(1965年諾貝爾物理學獎獲得者)曾經說過"What I cannot create, I do not understand.”(我不能理解我不能創造的東西)。對于生物無機化學的重要內容—金屬酶模擬,利用簡單的合成模型分子重構金屬活性部位,“自下而上”地重現天然金屬酶的功能,是理解和揭示金屬酶的構-效關系的重要手段,為金屬酶研究提供重要理論和實驗依據。按照該研究范式,我們課題組開展針對天然鎳葉綠素的研究工作,首次提出它在光合作用系統中可能扮演的電子傳遞/催化的重要角色,為理解這一自然界罕見的金屬輔酶提供了重要線索,同時也為設計非貴金屬能源催化劑提供了新視角。鎳葉綠素(Tunichlorin)首次由美國Rinehart教授于1989年從加勒比海的被囊生物Trididemnum soidum中分離并確認結構。如圖1所示,鎳葉綠素是一種以金屬鎳為中心的二氫卟吩輔酶,以焦脫鎂葉綠酸作為配體,卟啉環的β位具有一個不常見的羥甲基取代基,是一類罕見的葉綠素類金屬輔酶。迄今為止,自然界中含鎳離子的四吡咯輔酶有兩類。一類是輔酶F430, 是以鎳為中心的corphin結構,已被證實是一類催化甲烷轉化的金屬輔酶,其結構和在氧化還原過程中的電子傳遞功能較為明確。另一類為鎳葉綠素,但由于其結構不穩定,確切的功能還未知。Rinehart教授等推測,在海鞘的光合作用系統中,氫化酶中鎳簇中心分解后,鎳離子取代葉綠素中的鎂離子生成鎳葉綠素,因此鎳葉綠素的形成被看作是光合作用系統中鎳簇氫化酶中心的代謝方式。“鎳葉綠素是否僅僅作為鎳離子的“回收站”,還有沒有其它的功能?” 針對此問題,我們課題組基于鎳卟啉和F430輔酶在氧化還原反應中的研究結果,開展了模擬Tunichlorin結構,探索其獨特的葉綠素結構和催化產氫過程中電子傳遞功能的關系。我們的前期工作研究了葉綠素結構中的吡咯環飽和度對鎳離子反應性的影響(Chem. Sci., 8, 5953-5961, 2017;highlighted by Nat. Rev. Chem. 1, 0062, 2017)。我們發現吡咯環還原加氫后,卟吩共軛大環的飽和度增加,環構象柔韌性加大,表現出較強的穩定低價金屬的能力。鎳二氫卟吩卟啉應用于電催化析氫反應(HER)的活性提高約25倍。另一方面,天然鎳葉綠素中不尋常的β-羥基取代基引起了我們的興趣。在β-位引入羥基或羧基等取代基的四吡咯金屬輔基在自然界中廣泛存在,其β-位取代基的作用除了穩定飽和雙鍵外,還被認為在催化過程中扮演著組成次級配位環境并進行質子耦合電子轉移(PCET),調控反應的作用。因此,我們認為在鎳二氫卟吩模型化合物中引入β-羥基將會加深我們對鎳葉綠素的構效關系的認識,幫助我們破譯鎳(II)輔酶的功能。▲圖1. Macrocycle structures of natural Tunichlorin and the model complex described in this work.在此,我們利用前期發展的卟吩內酯化學作為分子平臺,從二氫卟吩內酯化合物出發,通過還原內酯基團得到具有β-位羥基的異菌綠素型鎳配合物。化合物的合成路線如圖2所示,由鎳卟啉型化合物1為起始,通過其β-位吡咯雙鍵的還原可以得到鎳二氫卟吩型化合物2。通過對其β-位吡咯雙鍵的內酯化可以得到鎳卟吩內酯型化合物3。將化合物3的內酯鍵還原氫化可以得到化合物鎳二氫卟吩型內半縮醛化合物4。在化合物3的基礎上繼續β-位還原可以得到鎳的二氫卟吩內酯化合物5,將5還原可以得到鎳異菌綠素型內半縮醛化合物6。對化合物6進行甲基化可以得到化合物7。化合物4和6的晶體圖如圖2b和c所示,都表現出相似的折疊的構象,其沿著N-Ni-N軸的扭轉角分別為33.5和34.5,Ni-N鍵的長度分別為1.923 ?和1.925 ?。▲圖2. 鎳配合物的合成路線(a)及化合物4(b)和化合物6(c)的晶體圖。化合物1-5和7在循環伏安圖中均表現出兩個可逆的還原電位(表1),第一還原電位在-0.99至-1.28V,第二還原電位在-1.59到-1.83V。與其他的鎳配合物不同,化合物6在-1.25V處有一個不可逆的還原峰。這個不可逆峰在掃速增加到200 mV s-1后會消失。而將化合物6的羥基甲基化后得到的化合物7,-1.25V處的可逆峰消失。以上現象表明這些不可逆峰是內半縮醛上羥基所導致的。我們計算出各個可逆還原電位電子傳遞的標準速率常數(ks),如表1所示,化合物1-3和5、7的電子傳遞速率都在0.02-0.03 cm s-1之間。而含有羥基的化合物4和6的第二還原電位的電子傳遞速率可以分別達到0.08和0.11 cm s-1。將化合物6的羥基甲基化后,化合物7電子傳遞速率常數降低為原來的0.03 cm s-1。該現象表明,β-位的羥基化在對電子傳遞速率的快慢有很大影響。
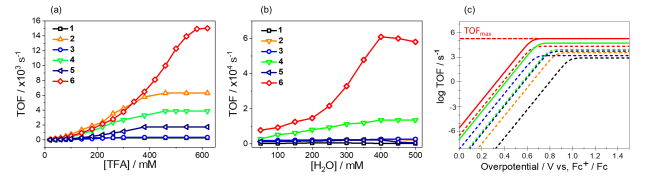
a. 所有的循環伏安實驗均使用三電極系統,掃速為0.1V s-1,以二茂鐵作為標準零電位(Fc+/Fc, E1/2 = 0.45 V vs. SHE)。Ni(II)卟啉類配合物的電催化HER活性總結在圖3和表2中。使用FOWA方法計算出化合物1-6在催化產氫反應中的反應速率TOF(表2)。我們發現,鎳異菌綠素型內半縮醛化合物6表現出最高的反應速率,其數值可達到1.5×104 s-1,與卟啉型化合物1相比,前者的TOF是后者的59倍。該結果表明,β-位羥基和不飽和增加在增加產氫反應的效果中都扮演著重要角色。對比含有羥基的化合物6的TOF與二氫卟吩內酯化合物5,前者是后者的9倍。同樣對比含有羥基的化合物4的TOF與卟吩內酯3,前者是后者的11倍。這說明了β-位羥基在析氫反應中的重要作用。有趣的是,在該體系中添加水(100至400 mM),化合物2、4和6的催化電流繼續抬升,而對于其他化合物來說,催化反應頻率的增加均不明顯(< 2倍)。我們將酸的種類變換為三乙胺鹽酸鹽(pKa = 10.0 in CH3CN)、甲磺酸(pKa= 1.6 in CH3CN)和對甲基苯磺酸(TsOH, pKa= 8.6 in CH3CN),均可以得到看到化合物6的催化電流能夠繼續上升。并且,在對甲苯磺酸為260mM的反應液中加入濃度為400mM的水,所有化合物的催化電流均有所增加。化合物6的催化電流可以從4.2 mA上升到10.0 mA,TOF從5.5×103達到6.1×104 s-1。對于同樣具有羥基基團的化合物4,在該條件下催化反應頻率也有5.4倍的增加。同樣的,我們對比無水體系和含400 mM水的體系化合物6的催化伏安曲線,可以看到,在含水體系中,催化反應的催化電勢一直保持在1.3 V,當酸的濃度達到100 mM時,催化電流能夠達到2.7 mA。而在相同酸濃度下,無水體系中的催化電流只有1.5 mA。以上結果表明,β-位羥基基團能夠在水的作用下有效增加析氫反應速率。▲圖3.((a)反應速率TOF與三氟乙酸濃度的關系圖,(b)反應速率TOF與水的濃度關系圖。(c)化合物1-6的催化Tafel曲線。
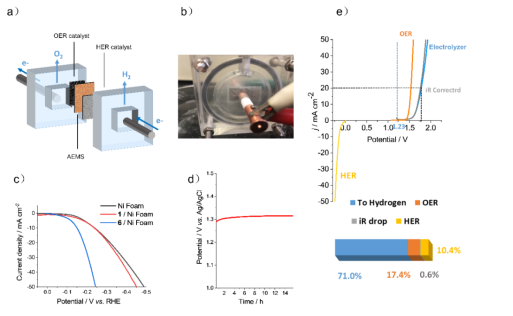
我們將化合物6擔載的泡沫鎳作為工作電極,與市售的RuO2組成雙電極系統,在如圖4a中所示的裝置中組成電解池。如圖4e中所示,該電解池的過電勢為η20 = 540 mV。對于組成電解池的兩個半反應來說,產氧反應的過電勢η20 = 300 mV,而產氫反應的過電勢為η20 = 182 mV。該電解池的過電勢比兩個半反應的過電勢要略高一些,高出的部分主要來自于電解池自身的內阻(iR drop)。由此可以計算出該電流密度下,電能轉化為氫能的效率為71.0%,其中由于OER過電勢產生的能量損失為17.4%,由HER造成的能量損失為10.4%,剩下的能量損失來自于電池自身內阻0.6%。▲圖4. 兩電極系統電解池的裝置示意圖(a)及其實際工作圖(b)。(c)0.5mg cm-2催化劑6和催化劑1分別擔載在泡沫鎳與沒有擔載催化劑的泡沫鎳作為工作電極的極化曲線的比較。(d)催化劑6作為工作電極的恒電流穩定性測試(e)電解池的極化曲線與與該工作條件下電解池的能量效率分析。
如圖5a所示,在施加-1.2V (vs. Fc+/Fc)電壓的條件下,化合物6得到一個電子變為[6]-,其紫外-可見吸收光譜的Soret band和Q-band強度均有下降,在514,704nm處先后出現新的吸收峰,說明卟啉環上形成了陰離子自由基。在施加-1.5V (vs. Fc+/Fc)電壓的條件下,化合物6得到兩個電子變為[6]2-,其Soret band繼續下降,其中384nm處的吸收峰比360nm處的吸收峰下降更明顯;704nm處的吸收峰繼續升高,但沒有由6變為[6]-時明顯。