本文作者:吳俊穎,余平,杜曼,等
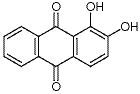
蕓苔素內(nèi)酯(brassinolide,1)是一種活性極高的植物內(nèi)源激素,化學(xué)名稱為(22R,23R,24S)-2α,3α,22,23-四羥基-24-甲基-B-高-7-氧雜-5α-膽甾-6-酮(見圖1)。蕓苔素內(nèi)酯能夠加快植物細(xì)胞伸長(zhǎng)和分裂,調(diào)節(jié)植物的光合作用,提高植物的抗逆性(抗寒、抗旱、抗鹽堿、抗病等),可施用于多種作物,在農(nóng)業(yè)生產(chǎn)中應(yīng)用前景廣闊。
圖1 蕓苔素內(nèi)酯的結(jié)構(gòu)式
蕓苔素內(nèi)酯存在于植物花粉、根、莖、葉和種子等多個(gè)部位,但含量極低,如花粉中含量約為1~100 ng/g(濕重)。因此,以天然存在的甾醇類似物為原料,人工合成蕓苔素內(nèi)酯成為了獲得蕓苔素內(nèi)酯的主要途徑。近幾十年來(lái),人們對(duì)蕓苔素內(nèi)酯的合成進(jìn)行了不斷的探索和研究,本文將對(duì)其合成方法進(jìn)行綜述。
1 蕓苔素內(nèi)酯的發(fā)現(xiàn)過(guò)程
1970年,Mitchell等首次報(bào)道在油菜花粉中發(fā)現(xiàn)了生物活性極高的物質(zhì),能引起菜豆幼苗節(jié)間伸長(zhǎng)、彎曲甚至裂開的異常生長(zhǎng)反應(yīng),但基于當(dāng)時(shí)的分離、鑒定手段,沒(méi)有確證結(jié)構(gòu)。1979年,Grove等從40 kg油菜花粉中提取到4 mg結(jié)晶,經(jīng)過(guò)XRD分析確定其結(jié)構(gòu)并命名為蕓苔素內(nèi)酯。
2 蕓苔素內(nèi)酯的合成路線
1980年,F(xiàn)ung等首次報(bào)道由豆甾醇(2)立體選擇性合成蕓苔素內(nèi)酯的方法。基于Salmond、Hutchins等報(bào)道的合成中間體3的方法,利用C-20手性將中間體3與E-構(gòu)型的鋰試劑(4)反應(yīng)合成化合物5,經(jīng)間氯過(guò)氧苯甲酸(MCPBA)氧化、LiBH4/BH3·THF還原,將C-24構(gòu)型翻轉(zhuǎn)得化合物7和8,區(qū)域選擇性為3∶1。(22R,23R,24S)構(gòu)型的化合物7經(jīng)酸催化重排、縮酮化、C-3羥基磺酰化、硼氫化-氧化引入C-6羥基、Li2CO3/DMAC體系消除、Jones氧化C-6羥基、四氧化鋨(OsO4)氧化C-2雙鍵、三氟過(guò)氧乙酸(CF3CO3H)將B環(huán)內(nèi)酯化并水解保護(hù)基合成蕓苔素內(nèi)酯(見圖2)。
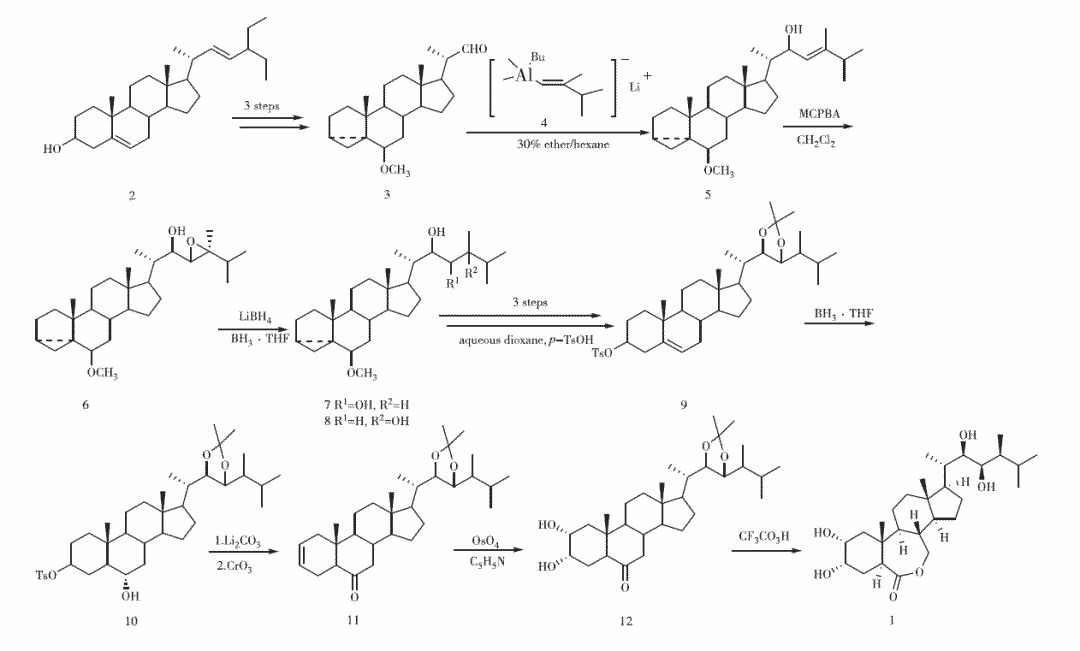
圖2 Siddall課題組的合成路線
同年,Ikekawa等以(3β,20S)-3-[(四氫-2H-吡喃-2-)氧基]孕甾-5-烯-20-醛(13)為起始原料與3-甲基-1-丁炔基鋰反應(yīng)合成化合物14(見圖3)。隨后經(jīng)還原、環(huán)氧化、反式開環(huán),得到(23R,24S)-24-氰基化合物(15),經(jīng)二異丁基氫化鋁還原并水解、乙酰化保護(hù)C-3 羥基、NaBH4還原C-24醛基、甲磺化、碘代和氫化三丁基錫還原合成C-24甲基化合物16。甲磺酸酯(17)經(jīng)硼氫化-氧化、過(guò)氧化氫氧化、PCC氧化、LiBr消除得到化合物11。隨后借鑒VanRheenen等報(bào)道的用OsO4/NMO(N-甲基嗎啉-N-氧化物)體系氧化雙鍵的方法,將化合物11氧化為cis-二羥基化物。經(jīng)計(jì)算由化合物13合成蕓苔素內(nèi)酯收率為2%。
1982年,Mori等報(bào)道化合物2經(jīng)磺酰化、溶劑化、Jones氧化、對(duì)甲基苯磺酸/環(huán)丁砜異構(gòu)化合成化合物20,后經(jīng)C-2雙鍵氧化、縮酮化、O3氧化、Me2S還原得化合物22。基于該課題組早期合成廚蟻(Pharaoh ant)性激素時(shí)報(bào)道的方法將化合物22與砜(23)連接,化合物24在Na/Hg/MeOH/EtOAc中消除、MCPBA氧化C-22雙鍵、30%HBr/AcOH體系反式開環(huán)、AcOH/H2O體系中乙酰氧基取代溴原子合成(22R,23R)化合物18,最后經(jīng)CF3CO3H將B環(huán)內(nèi)酯化并水解保護(hù)基合成蕓苔素內(nèi)酯(見圖4)。
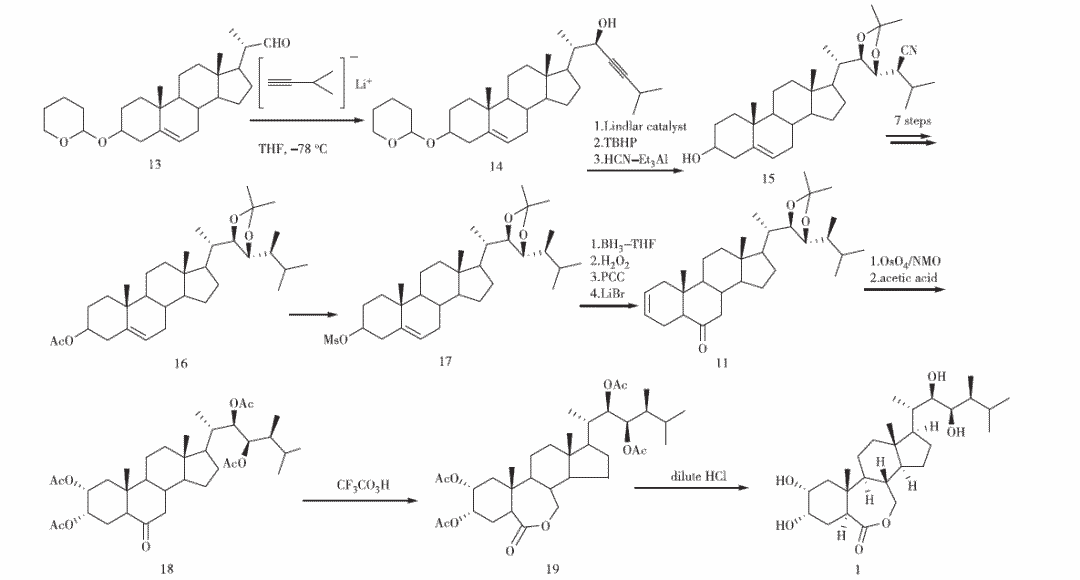
圖3 Ikekawa課題組的合成路線
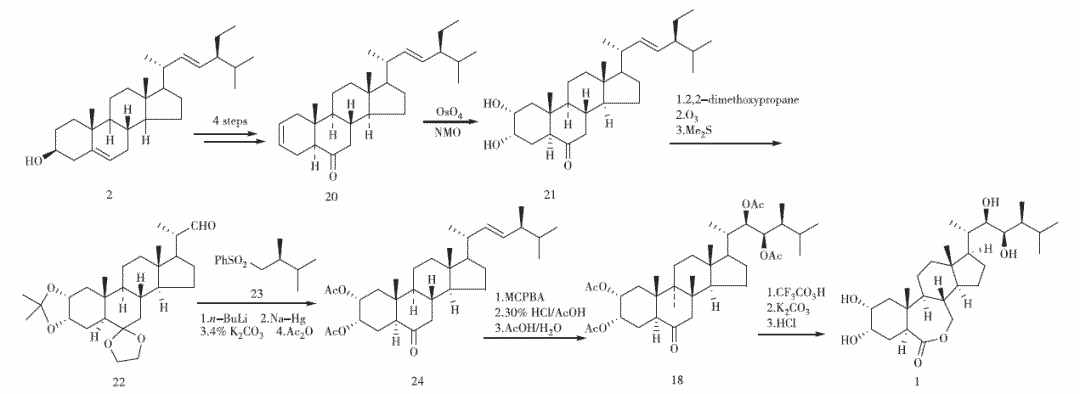
圖4 Mori課題組的合成路線1
1984年,Mori等報(bào)道了側(cè)鏈C-23、C-24環(huán)氧環(huán)的區(qū)域和立體選擇性開環(huán)方法。Ikekawa等曾嘗試在環(huán)氧環(huán)的C-24用有機(jī)銅試劑引入甲基未獲成功,Mori在構(gòu)建側(cè)鏈時(shí)用Me3Al開環(huán),實(shí)現(xiàn)了直接在C-24位接上甲基,得到構(gòu)型正確的蕓苔素內(nèi)酯的側(cè)鏈(見圖5)。異丙基炔基鋰(25)對(duì)化合物22的醛基加成、P2-Ni/H2/H2NCH2CH2NH2還原叁鍵、MCPBA環(huán)氧化雙鍵、Me3Al/n-BuLi開環(huán)得化合物27。由化合物2經(jīng)16步反應(yīng)合成蕓苔素內(nèi)酯,總收率3%。該方法具有很好的立體選擇性,但Me3Al試劑用量大且不穩(wěn)定,反應(yīng)需在-70 ℃、Ar保護(hù)下反應(yīng)69 h。
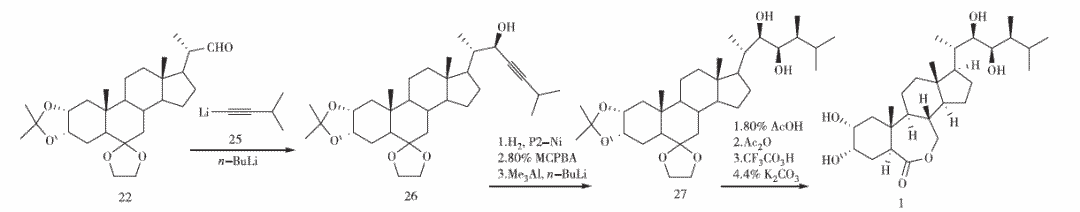
圖5 Mori課題組的合成路線2
1987年Mori等又報(bào)道了利用新型中間體i-甾酮二乙酰化物(30)合成蕓苔素內(nèi)酯的方法(見圖6)。其中化合物31是由化合物28經(jīng)Pd-C/H2脫芐醚等4步得到,比甲基醚化合物經(jīng)6步轉(zhuǎn)化成烯酮收率高且步驟少。化合物31經(jīng)雙鍵氧化、羥基保護(hù)后以77%的收率得到化合物32。全程共15步,總收率為9%。
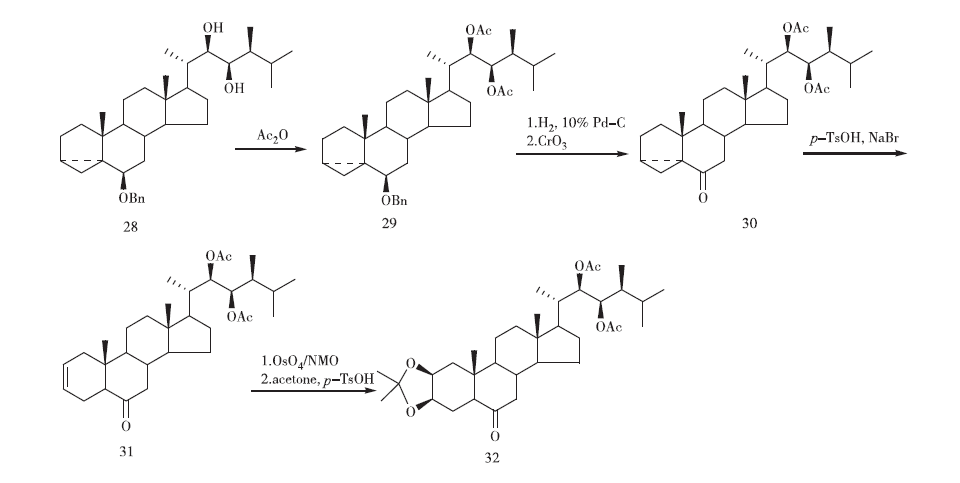
圖6 Mori課題組的合成路線3
1991年,Sun等一改以前先環(huán)氧化再開環(huán)的方法,用OsO4催化化合物33側(cè)鏈C-22雙鍵不對(duì)稱雙羥化,使反應(yīng)步驟簡(jiǎn)潔,但具有(24S)-24-甲基的類固醇主要產(chǎn)生(22S, 23S)-異構(gòu)體。該課題組基于Yamamoto和Sharpless的研究,發(fā)現(xiàn)在OsO4-K3Fe(CN)6-DHQD體系中完成雙鍵氧化可得到25%的(22R,23R)構(gòu)型的蕓苔素內(nèi)酯(見圖7)。
同年,McMorris等發(fā)現(xiàn)甾體母核與丁烯內(nèi)酯鋰鹽發(fā)生Aldol縮合時(shí),主產(chǎn)物構(gòu)型遵守Cram規(guī)則即為(22R)構(gòu)型,C-23的構(gòu)型則由縮合時(shí)的反應(yīng)溫度決定,低于-70 ℃時(shí)以(R)構(gòu)型為主。化合物3在-78 ℃/LDA條件下與3-異丙基丁烯內(nèi)酯(35)縮合得(22R, 23R)中間體36,其經(jīng)Pt/C催化氫化得到C-24異構(gòu)體比例(S∶R)=78∶22的混合物,化合物37經(jīng)LiAlH4還原側(cè)鏈內(nèi)酯、縮酮化、重鉻酸吡啶鹽(PDC)氧化端基醇、三(三苯基膦)氯化銠脫羰基得到(24S)甲基構(gòu)型產(chǎn)物41。化合物41經(jīng)p-TsOH/丙酮-水合成化合物42(見圖8),化合物42經(jīng)磺酰化合成化合物9,進(jìn)而合成蕓苔素內(nèi)酯。化合物3與2,化合物3-二甲基丁烯內(nèi)酯鋰鹽(43)縮合即得化合物44(見圖9),化合物44經(jīng)催化氫化、LiAlH4還原、MsCl/吡啶異構(gòu)化、LiAlH4還原伯醇得化合物46,化合物46經(jīng)縮酮化保護(hù)合成化合物42。由化合物35構(gòu)建側(cè)鏈步驟較多,但Aldol縮合時(shí)產(chǎn)物構(gòu)型單一,2種側(cè)鏈構(gòu)建方法的總收率均在32%~34%之間(以化合物3計(jì))。
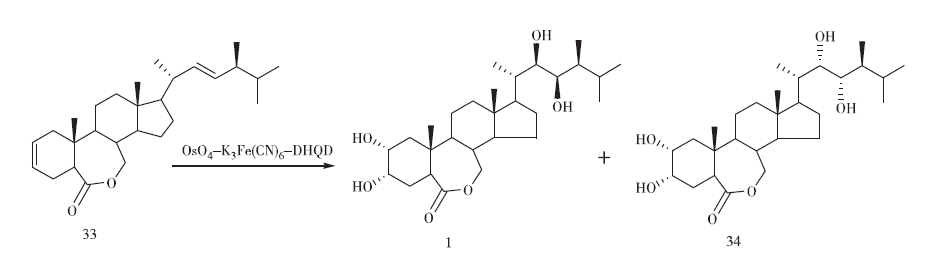
圖7 Zhou課題組的合成路線
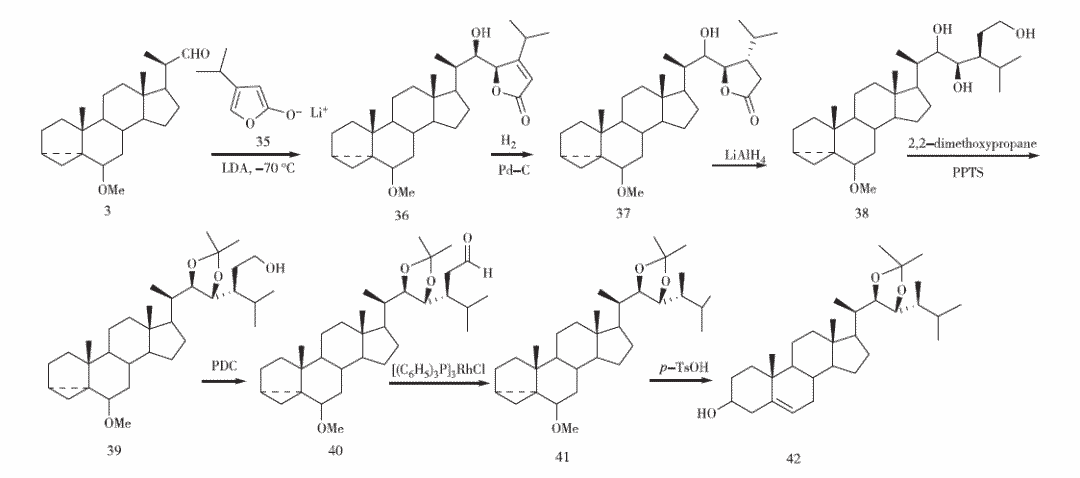
圖8 McMorris課題組的合成路線1
 圖9 McMorris課題組的合成路線2
圖9 McMorris課題組的合成路線2
1992年,Tsubuki等利用二氫吡喃酮中間體46構(gòu)建側(cè)鏈(見圖10)。縮合主產(chǎn)物48經(jīng)NBS/THF體系內(nèi)半縮醛化、PCC/CH2Cl2氧化、CeCl3/NaBH4/CH3OH-CH2Cl2還原C-23羰基、乙烯基乙醚/對(duì)甲苯磺酸吡啶鎓鹽(EE/PPTs)體系保護(hù)其C-23羥基、二甲基銅酸鋰/四氫呋喃[(CH3)2CuLi/THF] 與雙鍵加成、LiAlH4/乙醚體系還原、甲磺酰化、LiAlH4還原、水解得化合物51。
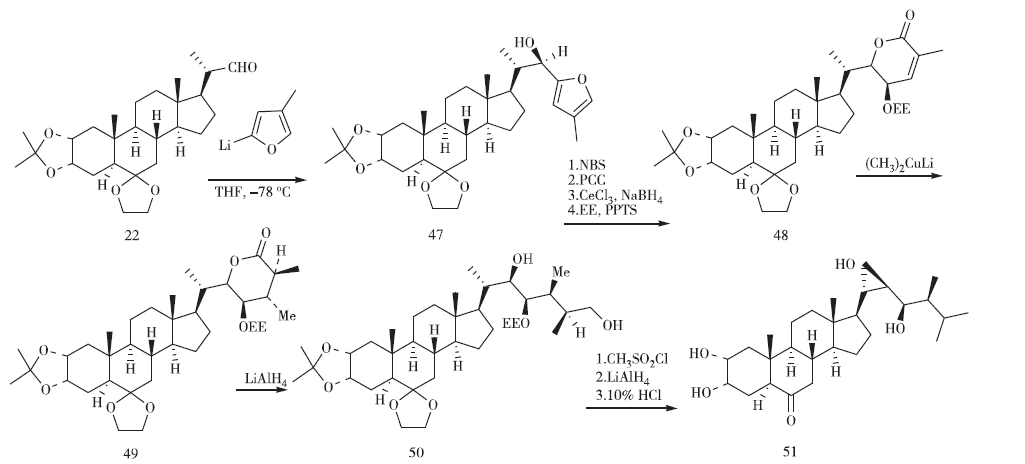
圖10 Honda課題組的合成路線
1994年,Hazra等采用了一條新的合成側(cè)鏈的路線(見圖11)。以醋酸妊娠雙烯醇酮52(16-DPA)為原料,經(jīng)C-16雙鍵選擇性催化加氫、C-3脫保護(hù)基、上保護(hù)、LiAlH4還原酮、POCl3/Py體系脫水得到C-17,C-20 cis-烯烴54。分別用三異丙基氯化鈦[Ti(i-Pr)3Cl]、三甲基氯硅烷[(CH3)3SiCl]和TBS-CL 3種催化劑對(duì)其進(jìn)行立體特異性烯反應(yīng),均生成(R)構(gòu)型產(chǎn)物55。化合物55經(jīng)MeOH/Py回流、Pd/C催化加氫、KOH/MeOH脫保護(hù)、CH3COOK/PCC/CH2Cl2氧化得化合物3,全程共11步,總收率為36%。化合物3經(jīng)1,3-二噻烷/正丁基鋰還原、乙酰化、NBS/BaCO3/Me2CO體系脫除二噻烷、Wittig反應(yīng)得化合物57,化合物57經(jīng)MCPBA/Na2HPO4將雙鍵環(huán)氧化、p-TSA/dioxane 將3,5位開環(huán)、(CH3)3Al/n-BuLi將環(huán)氧環(huán)開環(huán)并完成24位甲基化得46。該路線原料52較易得,但總的路線長(zhǎng),總收率為16%。
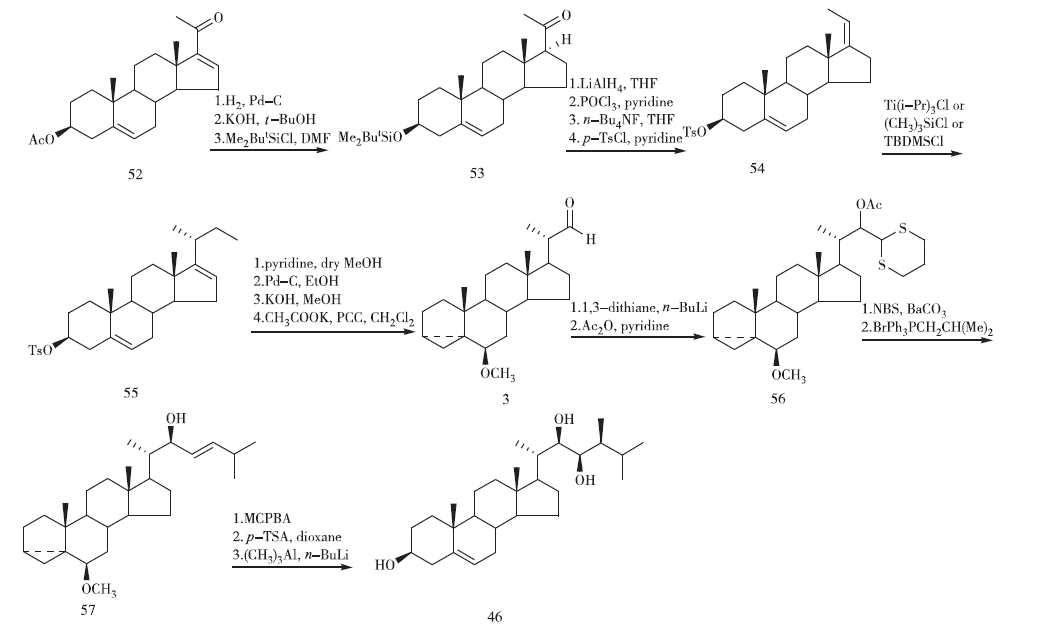
圖11 Hazra課題組的合成路線1
1997年,Back等采用了化合物22與金屬有機(jī)硒化合物58縮合(見圖12)構(gòu)建側(cè)鏈的方法。縮合得到含硒的C-23立體異構(gòu)體的混合物,且沒(méi)有相應(yīng)的C-22差向異構(gòu)體。將此粗品直接氧化,使原位硒氧化物順式消除,主要轉(zhuǎn)化為trans-烯丙醇(60)。化合物60在過(guò)氧化羥基異丙苯/鈦酸四異丙基酯/L-(+) 酒石酸二乙酯體系中環(huán)氧化、蘇式環(huán)氧化物經(jīng)異丙基氯化鎂/氰化亞銅體系開環(huán)得化合物27。由化合物2合成蕓苔素內(nèi)酯共12步,總收率為8%。
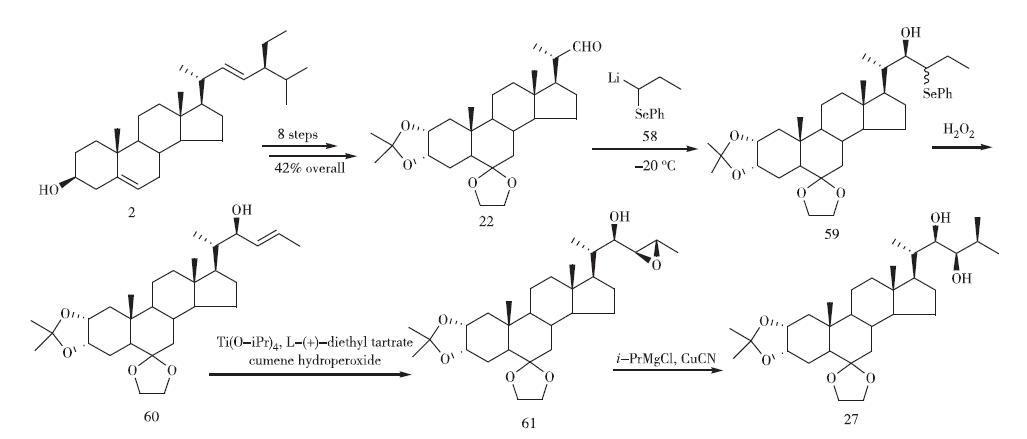
圖12 Back課題組的合成路線
2003年,Peng等也是采用甾體母核和側(cè)鏈連接的方式合成蕓苔素內(nèi)酯,在Aldol縮合中化合物3可以和α-甲硅烷氧基酮(62)在-78 ℃ LDA/THF體系下反應(yīng)(見圖13),也可以在TiCl4的介導(dǎo)下,使用硅基烯醇醚(66)代替烯醇化鋰鹽反應(yīng),化合物63脫除硅基給出(22R,23R)構(gòu)型的中間體64。在化合物3和側(cè)鏈62連接時(shí),若溫度為-78 ℃且在此溫度下淬滅反應(yīng),則生成(22R,23R)的中間體。若溫度為-78 ℃而淬滅溫度高于0 ℃,則生成(22R,23S)的異構(gòu)體,需要進(jìn)行C-23異構(gòu)化。化合物64經(jīng)縮酮化、Wittig反應(yīng)、催化加氫、脫保護(hù)基得到比例為70∶30的化合物46和68的混合物,化合物46可依圖9的方法合成蕓苔素內(nèi)酯,而化合物68是合成24-表蕓苔素內(nèi)酯的重要中間體。
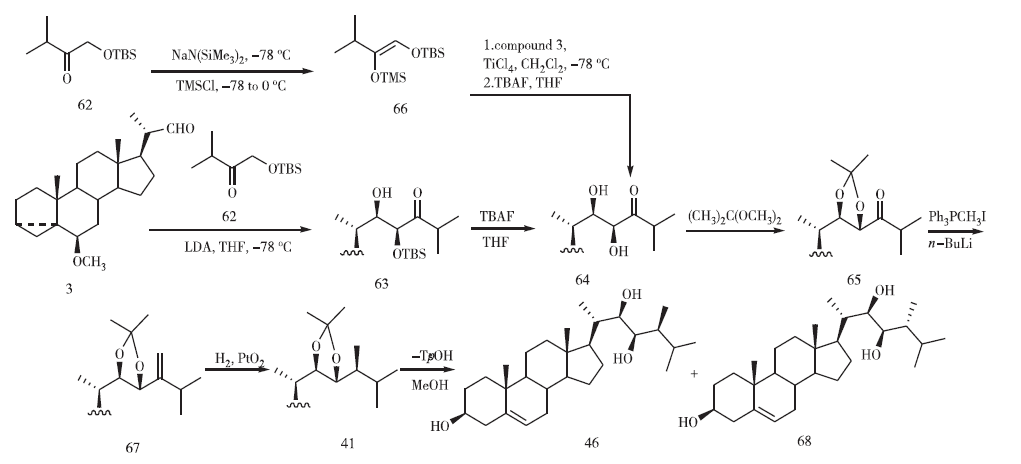
圖13 Li課題組的合成路線
2013年,Hurski等提出了以具有較大空間位阻和能有效穩(wěn)定碳負(fù)離子的硫縮醛(69)作為合成子與甾核偶聯(lián),隨后經(jīng)NaClO2/2-甲基-2-丁烯/NaH2PO4體系構(gòu)成酮、TBSOTf/2,6-二甲基吡啶體系保護(hù)C-22羥基,防止C-22羥基螯合控制,用二異丁基氫化鋁(DIBAL-H)還原再用TBAF脫C-22羥基保護(hù)基構(gòu)建側(cè)鏈(見圖14)。
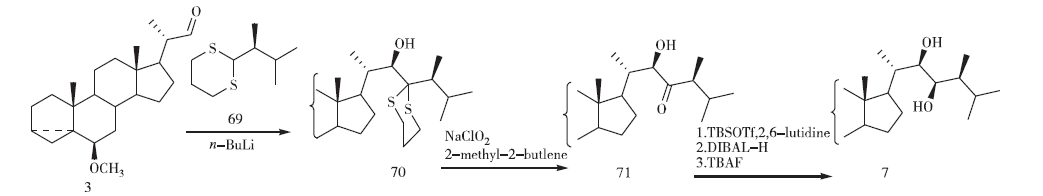
圖14 Zhabinskii課題組的合成路線
3 結(jié)論
蕓苔素內(nèi)酯的合成已經(jīng)取得了極大的進(jìn)步,但在其合成中仍然存在反應(yīng)步驟多、反應(yīng)體系復(fù)雜、溶劑用量大以及操作比較復(fù)雜等不利因素,所使用的部分試劑存在毒性大、易造成環(huán)境污染等問(wèn)題,特別是手性異構(gòu)體的生成導(dǎo)致收率下降。因此,探索更加適于工業(yè)生產(chǎn)的合成方法和新的立體專一性合成路線以滿足市場(chǎng)需求,仍是研究人員努力的方向。
來(lái)源: 《農(nóng)藥》雜志