合成脂肪族硝基化合物最便捷的方法是鹵素,磺酸酯等的硝基取代。活潑亞甲基的硝化,烯烴的硝化,和胺、肟的氧化,也能合成脂肪族硝基化合物。
1 硝基取代反應(yīng)示例
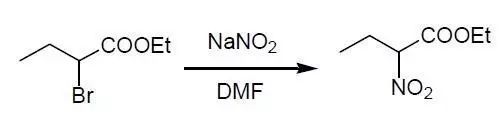
鹵代烴與亞硝酸鹽(NaNO2,KNO2,AgNO2等)在極性非質(zhì)子溶劑DMF或DMS0中或在相轉(zhuǎn)移催化條件下反應(yīng),可以得到較好產(chǎn)率的硝基化合物。通常反應(yīng)伴有副產(chǎn)物亞硝酸酯的生成。磺酸酯類化合物,如Tosylate,也可以發(fā)生類似反應(yīng)。這是合成脂肪族硝基化合物的最常用的方法。
三甲硅基以及三甲錫基也能被硝基取代,得到相應(yīng)的硝化物。
Organic Syntheses, Coll. Vol. 4, p.454; Vol. 37, p.44
Ethyl α-bromobutyrate (58.5 g., 0.30 mol) is poured into a stirred mixture of sodium nitrite (36 g, 0.52 mol), anhydrous phloroglucinol (40 g, 0.32 mole), and 600 mL of N,N-dimethylformamide (DMF) contained in a 1-L three-necked flask equipped with a sealed stirrer. The flask is closed, except for a tube containing calcium chloride, and immersed in a water bath maintained at room temperature. Stirring is continued for 2.5 hours; then the reaction mixture is poured into ice water (1.2 L) layered over with ether (300 mL) After separation of the upper layer, the aqueous phase is extracted with four 100-mL portions of ether. The combined extracts are washed with four 100-mL portions of water and then dried
over anhydrous magnesium sulfate. The magnesium sulfate is removed by suction filtration and washed with four 25-mL portions of ether which are combined with the filtered extract. The ether is distilled through a small column, under reduced pressure, from a 1-L flask which is heated by a bath whose temperature is gradually raised to about 60°. The residual yellow liquid is transferred, with the aid of a little anhydrous ether, to a 100-mL flask, and the
remaining solvent is distilled through the column under reduced pressure. Rectification of the residue yields 2–3 g of fore-run boiling in the range 33–71°/1 mm. which is followed by (33–36 g, 68–75%) of colorless ethyl α-nitrobutyrate, bp 71°/1 mm., nD20 1.4233.
2 含活潑亞甲基化合物的硝基取代反應(yīng)示例
含活潑亞甲基(如芐甲基,含α氫的酯等)的化合物,在強(qiáng)堿(K/NH3, Na/NH3)的作用下,與硝酸酯反應(yīng),得到硝基化合物。
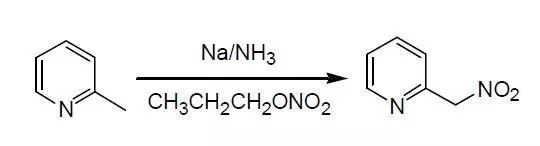
Feuer, H; Lawrence, J. P. J. Org. Chem. 1972, 3662.
To a freshly prepared solution of sodium amide (0.23 mol) in liquid ammonia (300 Ml) was added 2-picoline ((8.4 g, 0.09 mol)) rapidly at –33o. After stirring for 10 min, n-propyl nitrate (29.6 g, 0.28 mol) was added as rapidly as possible while the temperature was kept below –33o. The mixture was stirred an additional 5 min, the ammonia gradually replaced with absolute ether, and the reaction mixture filtered after room temperature was reached (3-5hr). The crude sodium 2-picolylnitronate was dried in vacuo, dissolved in 20 mL of water,
and acidified with 11.0 g of glacial acetic acid at room temperature. Extracting the solution with chloroform, drying (Na2SO2), concentrating the extract in vacuo, and distilling the residue afforded 2-nitromethylpyridine (7.3 g, 55%).
3 胺和肟的氧化反應(yīng)示例
胺的氧化,特別是肟的氧化,是合成脂肪族硝基化合物的一個(gè)重要手段。因?yàn)殡靠?span style="line-height: 1.6;">以方便地由醛酮和羥胺制得。這是由醛酮合成硝基化合物的有效的方法,常用的氧化劑有過酸、臭氧、Oxone,高錳酸鉀,高硼酸鈉,次氯酸。
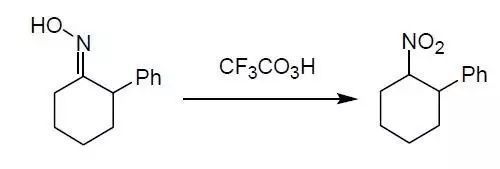
Sundburg, R. J; Bukowich, P. A. J. Org. Chem. 1968, 33, 4098.
A mixture of oxime (1.89 g, 10 mmol) , urea (0.2 g, 3 mmol), NaH2PO4 (7.8 g, 54 mmol) in acetonitrile (25 mL) was stirred and heated to gentle reflux. A solution of
peroxytrifluoroacetic acid was prepared by adding, dropwise during 10 min, trifluoroacetic anhydride (3.4 mL, 24 mmol) to a solution of 90% hydrogen peroxide (0.55 mL, 20 mmol) and acetonitrile (6 mL) chilled in ice bath. The peroxytrifluoroacetic acid solution was then added dropwise over 1 h to the stirred, heated oxime miture. In most cases, the mixture turned blue or green as the oxidizing solution was added but became yellow by the time addition was completed. The reaction mixture was then centrifuged, and the yellow supernatant liquid was decanted and concentrated in vacuo. The residue was treated with water (15 mL) and then extracted with 3 portion (15 mL each) of methylene chloride. The combined organic extracts were washed with 5% aqueous sodium bicarbonate, dried over MgSO4, and concentrated residue to give the product (1.95 g, 95%).
4 烯烴硝化反應(yīng)示例
烯烴直接硝化生成共軛硝基取代烯烴,是一個(gè)合成脂肪族硝基化合物的有效的方法。常用的有 NO,N2O4,NaNO2/CAN, NaNO2/HgCl2 等多種體系。
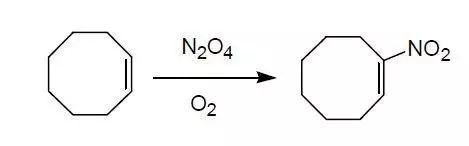
Organic Syntheses, Coll. Vol. 6, p.837; Vol. 50, p.84.
Sodium-dried diethyl ether (150 mL) is placed in a 1-L, four-necked flask equipped with a fritted gas-inlet extending to its bottom, a sealed mechanical stirrer, a 100-mL, pressure-equalizing dropping funnel, a thermometer, and a dry ice condenser protected with a phosphorus pentoxide drying tube. Cyclooctene (44.4 g, 53.5 mL, 0.404 mol) is placed in the dropping funnel, and the system is swept with dry oxygen. Dinitrogen tetroxide (39.3 g, 27.1mL at ?9°, 0.427 mole) is condensed in a graduated, calibrated trap that is protected with a phosphorus pentoxide drying tube and has been swept with dry oxygen.
The flask is cooled to ?10°, and the dinitrogen tetroxide is distilled with a warm water bath from the trap into the ether, with slow stirring; the transfer is aided by a minimal flow of dry oxygen. The solution is allowed to warm to 0–5°, and the oxygen flow rate is increased to 10 mL per minute. The cyclooctene is dropped into the dinitrogen tetroxide solution, with vigorous stirring, over a 30-minute period. The reaction is exothermic, and the temperature is kept at 9–12° by cooling with a methanol–dry ice bath at ?20°. The dropping funnel is rinsed
with 25 mL of ether, and the yellow solution is stirred for an additional 30 minutes at 10° with continued oxygen flow. Triethylamine (121 g, 1.20 mol) is added, with stirring, over a 12-minute period; the temperature of the reaction mixture is kept at 4–12° by maintaining the bath at ?4°. The mixture is kept at room temperature for an additional 30 minutes, diluted with 150 mL of ether, and cooled to 0–5°. The excess triethylamine is neutralized with an ice-cold solution of 72 g of acetic acid in 200 mL of water, with stirring. The reaction mixture is transferred to a 2-l. separatory funnel and extracted with three 400-mL portions of ether.
The combined ethereal extracts are washed with two 200-mL portions of water, three 150-mL portions of saturated aqueous sodium hydrogen carbonate, and again with water. Most of the ether is removed at room temperature with a rotary evaporator. The water that separates is removed with the aid of a small separatory funnel, and the remaining ether, traces of water, and cyclooctane are distilled at room temperature (10 mm) over 3 hours, yielding 59–61 g of the crude 1-nitrocyclooctene as a yellow oil and. Chromatography on silica gel
with successive elution with n-hexane and benzene gives 1-nitrocyclooctene (39–40 g,63–64%). Distillation gave an analytically pure sample, bp 60° (0.2 mm.), nD20 1.5116.
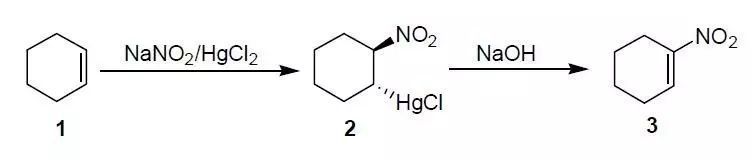
E.J.Corey, H. Estreicher, JACS, 1978, 6294.
Cyclohexene (0.82 g, 10 mmol) was added to the aqueous solution of mercuric chloride (2.71 g, 10 mmol) and sodium nitrite (1.38 g, 20 mmol) and stirred at 25oC for 30 h. The resulting precipitate was filtered to give the nitromercurial compound 2 (2.91 g, 80%). Compound 2 in methylene chloride (30 mL) was added 2.5 N aqueous sodium hydroxide (3.2mL, 8 mmol) at 25oC, the mixture was stirred for 5 min followed by acidification (1 N hydrochloric acid), the resulting mixture was filtered through Celite (quantitative recovery of metallic mercury), extracted with methylene chloride, the solvent was removed under reduced pressure to give pure l-nitrocyclohexene (1 g, 98%).
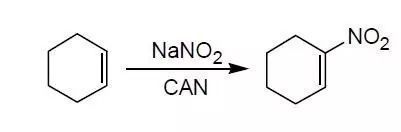
J. R. Hwu, K. L. Chen, S. Ananthan, J. Chem.Soc. Chem. Commun, 1994, 1425.
Cyclohexene (0.82 g, 10 mmol) was added to the mixture of ammonium cerium (IV) nitrate (NH4)2Ce(NO3)6 (CAN, 11 g, 20 mmol), and sodium nitrite (6.9 g, 100 mmol) and acetic acid (7.2 g, 120 mmol) in chloroform (100 mL) in a sealed tube. After sonication (600W) at 50oC for 4 h, the resulting mixture was washed with water and brine, extracted with chloroform. The solvent was removed under reduced pressure, the residue was purified by coumn chromatography to give the product (1.2 g, 95%).
本文內(nèi)容來源于網(wǎng)絡(luò),版權(quán)歸原作者所有