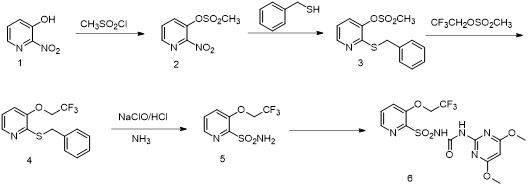
三氟啶磺隆可以由中間體3-(2,2,2-三氟乙氧基)吡啶-2-磺酰胺和2-氨基-4,6-二甲基嘧啶偶聯得到。后者可以大量得到,前者目前有3條合成路線。
合成路線一是選用2-氯-3-羥基吡啶為起始原料,首先選用芐基保護羥基,然后和硫脲反應,接著通入氯氣氧化再氨化,最后在鈀碳的催化下氫解脫保護得到3-三氟乙氧基吡啶-2-基磺酰胺。
合成路線二同樣是選用2-氯-3-羥基吡啶為起始原料,首先與對甲苯磺酸三氟乙酯反應,再與硫脲反應得到2-巰基-3-三氟乙氧基吡啶,通入氯氣后氨解得到3-三氟乙氧基吡啶-2-基磺酰胺。
合成路線三是以煙酰胺為起始原料,首先發生Hofmann降解得到3-氨基吡啶,然后被過氧化氫氧化后氯代得到2-氯-3-氨基吡啶,重氮化后再用三氟乙醇醇解得到2-氯-3-三氟乙氧基吡啶,再與硫氫化鈉反應得到2-巰基-3-三氟乙氧基吡啶,用次氯酸鈉氧化得到3-三氟乙氧基吡啶-2-基磺酰胺。
3條路線相比,合成路線一選用芐基保護羥基,而且脫保護要用貴金屬催化,后兩條路線步驟較短,不用保護羥基,直接反應。
在已有的合成路線的基礎上,筆者同樣選用不保護羥基的方法,以2-硝基-3-羥基吡啶為起始原料,首先和甲磺酰氯反應,然后和芐硫醇發生親核取代,和2,2,2-三氟乙基甲磺酸酯發生醚交換,再經過次氯酸鈉的氧化,氨氣氨化得到目標中間體,最后和2-氨基-4,6-二甲基嘧啶偶聯得到三氟啶磺隆。
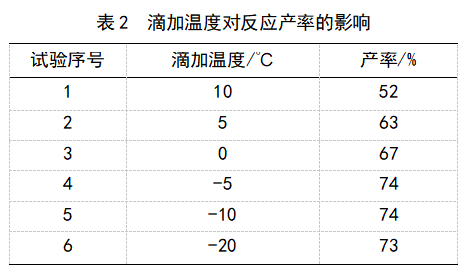
圖1 三氟啶磺隆的合成路線 1 試驗部分 1.1 主要儀器與試劑 Bruker AVANCE (核磁共振波譜儀600 MHz) 和Agilent 400 MHz;maXis超高分辨飛行時間質譜儀(maXis)。 所用試劑無特殊說明均為市售分析純試劑,由國藥試劑或薩恩化學技術(上海)有限公司提供。 1.2 三氟啶磺隆的合成 1.2.1 2-硝基吡啶-3-基甲磺酸鹽的合成 在50mL的茄形瓶中加入2-硝基-3-羥基吡啶(1.4g,10mmol)、三乙胺(1.39mL,1.3eq)和10mL二氯甲烷,將所得溶液冷卻至0 ℃,然后緩慢滴加甲磺酰氯(1mL,1.3eq)。滴加完畢后,溫度升高至室溫反應1.5h,反應結束后加入少量稀HCl和NaHCO3溶液洗滌,用乙酸乙酯萃取,得到的有機相用無水硫酸鈉干燥后旋干,得到化合物1為深黃色油狀物,質量為2.02g,不作進一步提純。 1.2.2 2-(芐硫基)吡啶-3-基甲磺酸鹽的合成 在50mL的茄形瓶中加入化合物1(2.02g)、芐硫醇(1.29mL,1.1eq)和15 mL N,N-二甲基甲酰胺,冰浴下攪拌。將氫氧化鉀(1.9g,1.4eq)溶解于4.4 mLH2O中制備30%的氫氧化鉀水溶液,冰浴下將氫氧化鉀溶液緩慢滴加至上述溶液中。滴加完畢后繼續冰浴反應1h,反應結束后將混合液倒入鹽水中,用乙酸乙酯萃取,得到的有機相用無水硫酸鈉干燥后旋干,柱層析得到化合物2為淡黃色固體,質量為1.51g,前兩步總產率為51%。試驗數據:1HNMR (400MHz, CDCl3) δ 8.41 (dd, J = 4.7, 1.4 Hz, 1H), 7.58 (dd, J = 8.1, 1.4 Hz, 1H), 7.38 (d, J = 7.2 Hz, 2H), 7.32 – 7.26 (m, 2H), 7.25 – 7.23 (m, 1H), 7.08 (dd, J = 8.1, 4.8 Hz, 1H), 4.47 (s, 2H), 3.17 (s, 3H). 1.2.3 2-(芐硫基)-3-(2,2,2-三氟乙氧基)吡啶的合成 在50 mL的茄形瓶中加入化合物2(1.48g,5mmol)、2,2,2-三氟乙基甲磺酸酯(1.6g,1.8eq)、碳酸鉀(1.03g、1.5eq)、10 mLN,N-二甲基甲酰胺,在120 ℃油浴下反應2 h。反應結束后,加入水淬滅,用乙酸乙酯萃取,得到的有機相用無水硫酸鈉干燥后旋干,柱層析得到化合物3為白色固體,質量為1.2 g,產率為80%。試驗數據:1HNMR (400 MHz, CDCl3) δ 8.20 (dd, J = 4.2, 1.9 Hz, 1H), 7.44 – 7.40 (m, 2H), 7.33 – 7.28 (m, 3H), 7.26 – 7.21 (m, 2H), 7.02 – 6.99 (m, 2H), 4.44 (s, 2H), 4.35 (q, J = 8.1 Hz, 2H)。 1.2.4 3-(2,2,2-三氟乙氧基)吡啶-2-磺酰胺的合成 在100mL的茄形瓶中加入化合物3(1.49g,5mmol)、15mL二氯甲烷、10 mL濃鹽酸,將所得溶液冷卻至-5 ℃,然后緩慢滴加有效氯含量為8%的次氯酸鈉(18.5mL,10eq),滴加完畢后室溫攪拌1.5h,反應結束后,用乙酸乙酯萃取,得到的有機層用無水硫酸鈉干燥,旋干后加入10mL二氯甲烷,冷卻至0 ℃,然后通入氨氣20min,旋干溶劑,柱層析得到化合物4為白色固體,質量為0.78 g,產率為61%。試驗數據:1HNMR (400MHz, DMSO) δ 8.26 (d, J = 4.4Hz, 1H), 7.83 (d, J = 8.1 Hz, 1H), 7.64 (dd, J = 8.5, 4.5 Hz, 1H), 7.31 (s, 2H), 4.99 – 4.91 (m, 2H).13CNMR (101MHz, dmso) δ 151.01, 148.63, 141.40, 128.53, 123.95, 122.65, 65.79。 1.2.5 三氟啶磺隆的合成 在50 mL的茄形瓶中加入化合物4(1.28g,5mmol)、4,6-二甲氧基-2-(苯氧基羰基)氨基嘧啶(1.6g,1.2eq)、10mL乙腈,冰浴降溫至0 ℃并緩慢滴加三乙胺(0.8mL,2eq),滴加完畢后室溫下攪拌1h,反應結束,旋干溶劑,殘液中加入少量稀HCl洗滌,然后用乙酸乙酯萃取,有機層用無水硫酸鈉干燥后柱層析得到化合物5,質量為1.88g,產率為86%。試驗數據:1HNMR (400MHz, DMSO) δ 12.75 (s, 1H), 10.60 (s, 1H), 8.31 (s, 1H), 7.88 (d, J = 8.6 Hz, 1H), 7.73 (d, J = 7.5 Hz, 1H), 5.96 (s, 1H), 4.99 (q, J = 9.1 Hz, 2H), 3.85 (s, 6H). 13CNMR (101MHz, DMSO) δ 172.04, 158.26, 157.79, 151.41, 150.89, 141.70, 127.18, 125.64, 81.43, 66.61, 54.33。 2 結果與討論 在該路線的合成步驟中,最為關鍵的是第4步化合物4的合成。這一步是氧化反應,選用的氧化劑是次氯酸鈉,首先芐硫基團和次氯酸鈉發生氧化還原反應,生成亞磺酸中間體,然后氯離子進攻取代得到磺酰氯,因此次氯酸鈉作為氧化劑需要過量,得到的中間體磺酰氯再被氨氣氨化得到化合物5。首先,在鹽酸加入量和反應時間不變時,于-5 ℃下改變次氯酸鈉的加入量,考察次氯酸鈉的用量對磺酰氯產率的影響,見表1。 試驗發現,隨著次氯酸鈉用量的增加,反應產率得到了提高,當次氯酸鈉的加入量是反應物的10倍時,生成磺酰氯的產率最高,達到74%,繼續提高次氯酸鈉的用量對反應產率的影響并不大。 由于這一步是氧化反應,在滴加次氯酸鈉過程中,會出現反應體系溫度升高的情況,一旦溫度升高,就會發生副反應。為了防止副反應對磺酰氯產率的影響,筆者采用低溫緩慢滴加的方法。在鹽酸、次氯酸鈉加入量和反應時間不變時,考察了滴加溫度對于磺酰氯產率的影響(表2)。 試驗發現,當滴加溫度高于0 ℃時,反應產率較低,可能有副反應發生,當溫度降低至-5℃以下時,能有效抑制副反應的發生,產率提高至74%。因此,最終選擇滴加溫度為-5℃。 筆者首先嘗試將得到的磺酰氯緩慢滴加至氨水,發現反應效果不佳,原料在長時間的反應條件下都不能反應結束,因此選擇將得到的磺酰氯溶解在二氯甲烷中,然后通入氨氣氨化,氨化的過程很快,效率很高,因此需要控制通入氨氣的時間。在恒定溫度為0 ℃時,通入不同時間的氨氣,考察氨化時間對磺酰氯產率的影響(表3)。 試驗發現,氨化時間為20 min時的效果最好,收率能夠達到82%,繼續延長反應時間,反而會使產物轉化為雜質,收率降低。 3 結論 本合成路線主要選用2-硝基-3-羥基吡啶為起始原料,首先經過兩步取代反應,然后發生醚交換,再氧化、氨化,最終偶聯得到產物三氟碇磺隆。本合成路線反應條件易于達到,可工業化應用。 來源:《世界農藥》第二期