▲第一作者:安倫,童非非 ;通訊作者: 張新剛 研究員
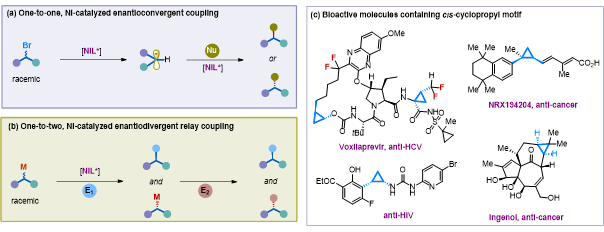
論文DOI:10.1021/jacs.0c04462本文首次提出了對映發散式不對稱接力偶聯(Enantiodivergent Relay Coupling, EDRC)的策略,并將該策略應用到鎳催化下消旋環丙基鋅試劑與1級,2級(氟代)烷基鹵代物的不對稱偶聯反應中,實現了“一鍋法”對映發散式合成具有不同構型、多種取代的手性三元環化合物。該反應為過渡金屬催化的不對稱偶聯反應提供了新思路,是首例過渡金屬催化下基于氟鹵烷烴的不對稱二氟烷基化反應。近年來,鎳催化的不對稱交叉偶聯反應作為一種立體選擇性高效構建碳-碳鍵的方法受到了化學家們的廣泛關注。加州理工學院的Gregory C. Fu教授在鎳催化下消旋親電試劑參與的不對稱偶聯領域做出了突出貢獻(Science 2017, 356, 152-160; ACS Cent. Sci. 2017, 3, 692-700)。該類反應中消旋親電試劑經烷基自由基中間體,在手性鎳催化劑的作用下與親核試劑發生手性歸一化偶聯反應,可以高立體選擇性生成立體構型單一的產物。相對于消旋親電試劑,鎳催化下消旋親核試劑參與的不對稱偶聯反應研究則相對空白(J. Am. Chem. Soc. 1976, 98, 3718-3719; J. Am. Chem. Soc. 2013, 135, 10946-10949; Angew. Chem., Int. Ed. 2017, 56, 5821-5824)。這一反應的主要難點是如何誘導消旋親核試劑生成手性烷基鎳中間體。雖然化學家們發展了手性親核試劑參與的立體專一性反應,但該類反應需要額外步驟制備光學純的親核試劑,并且很多手性親核試劑在反應過程中不穩定,易發生消旋化。因此,實現過渡金屬催化下消旋親核試劑參與的對映選擇性偶聯反應具有重要意義。同時,發展新的手性催化模式,在之前已發展手性匯聚式合成的基礎上,開發更加高效的手性發散式合成新策略也將為過渡金屬催化的不對稱偶聯反應注入新的活力。受到酶催化動力學拆分的啟發,作者設想利用手性鎳催化劑實現對消旋親核試劑的手性拆分,進而實現對映發散式不對稱接力偶聯反應(EDRC)。該過程可分為兩步,第一步反應在手性配體的誘導下,鎳催化劑能夠立體選擇性地拆分消旋親核試劑中一種構型底物與親電試劑反應得到光學純的偶聯產物,而另一構型的親核試劑在該步反應中不受影響。隨后,加入第二組份親電試劑捕獲第一步反應后剩余的另一構型親核試劑,從而可以生成具有不同取代,構型相反的產物。與傳統的鎳催化手性匯聚式合成方法相比(圖1a),該策略的特點在于能夠實現從單一消旋底物出發合成兩種具有不同取代,構型相反的手性化合物,具有高效簡潔,手性產物多樣的優點(圖1b)。該策略的關鍵在于如何高效實現第一步鎳催化的立體選擇性偶聯反應,并將第二步立體專一性偶聯反應串聯起來,做到一鍋高效合成。 考慮到手性三元環結構廣泛存在于醫藥分子和生物活性分子中(圖1c),作者選用了消旋三元環鋅試劑對EDRC反應進行了探究。▲圖1 圖片來源:J. Am. Chem. Soc.
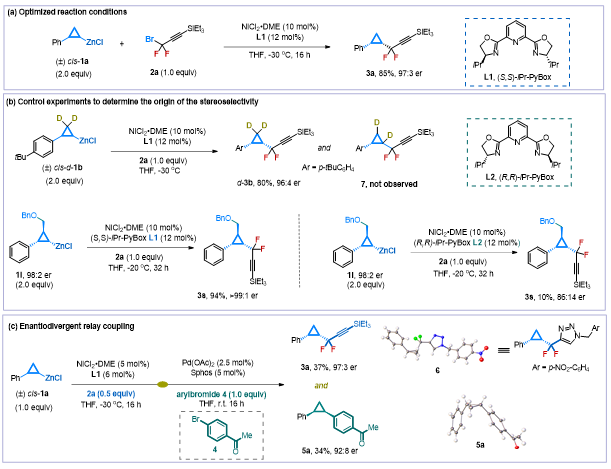
首先,作者對EDRC反應的第一步,鎳催化下消旋親核試劑參與的立體選擇性偶聯反應條件進行了探究。作者選用消旋的三元環鋅試劑 (±)-cis-1a為底物與偕二氟炔丙基溴代化合物2a進行偶聯,發現在NiCl2·DME/(S,S)-iPr-Pybox催化體系下能夠以85%的產率和97:3的對映選擇性生成產物3a (圖2a)。隨后,作者通過構型專一的鋅試劑1l,在不同構型手性配體的作用下證明了該反應是一個受手性配體控制的動力學拆分過程,并通過氘代實驗排除了β-H消除、遷移、插入的反應路徑(圖2b)。該立體選擇性偶聯反應為后續立體專一性偶聯反應奠定了重要基礎,進而作者對EDRC策略進行了驗證。如圖2c所示,從1.0當量的環丙基鋅試劑底物1a出發,在第一步鎳催化下與0.5當量的親電試劑2a發生立體選擇性的偶聯反應高效生成手性產物3a之后,作者向反應體系中加入了鈀催化劑(Pd(OAc)2/SPhos)和第二組份親電試劑4,通過立體專一性的偶聯反應高效高立體選擇性地生成了手性產物5a,其中第一步的鎳催化劑并沒有對第二步反應造成影響。X-ray單晶結構顯示,化合物3a和5a絕對構型相反,從而驗證了EDRC策略。▲圖2 圖片來源:J. Am. Chem. Soc.
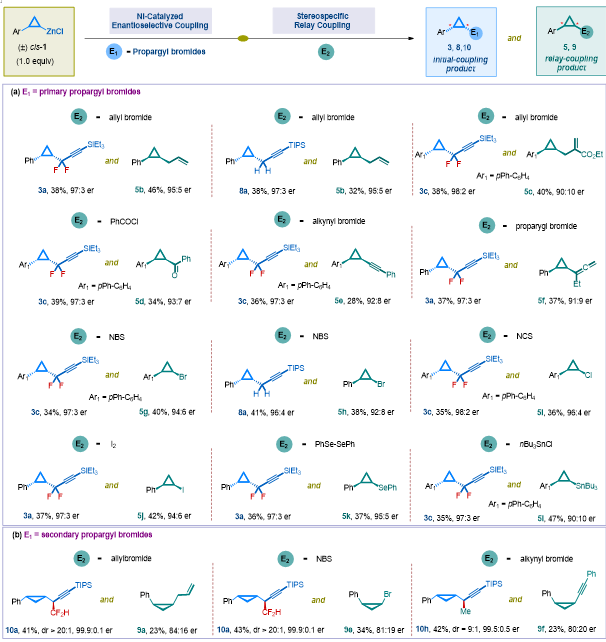
該鎳催化下消旋環丙基鋅試劑參與的不對稱偶聯反應,不僅拓展了消旋親核試劑參與的不對稱偶聯反應的應用范疇,同時也首次實現了過渡金屬催化下基于氟鹵烷烴的直接不對稱二氟烷基化反應。考慮到該步反應的重要性,在進行EDRC反應之前,作者首先對該反應進行了底物拓展。對環丙基鋅試劑1的考察表明,芳基上的許多重要官能團都可以得到較好的兼容(3a-3g)。并且,多取代的三元環鋅試劑也能夠順利進行反應(3h, 3i),實現一步構建三個連續手性中心,從而為合成多取代手性三元環化合物提供了一種高效方法。需要指出的是,烷基取代的偕二氟炔丙基溴代底物在反應過程中并沒有發生聯烯化副反應,體現了該反應優秀的區域選擇性(3j-3k)。一些復雜生物活性分子衍生的底物也能得到較好地兼容,表明該方法在合成包含手性三元環結構的生物活性分子中具有潛在應用價值(3l-3m)。由于手性三元環單元和二氟亞甲基(CF2)結構在生物活性分子設計中都具有重要應用,因此該方法也為立體選擇性合成二氟烷基取代的三元環化合物提供了一種高效簡便手段。該反應體系還可以進一步拓展到非氟底物。炔丙基溴代底物(8a-8e)和烯丙基溴化合物(9a-c)都表現出了良好的反應活性;即使是烷基碘代物也能給出良好的產率和立體選擇性(9d)。實現二級親核試劑和二級親電試劑的不對稱偶聯一直是手性合成中的一個挑戰性課題,因為反應同時會生成兩個手性中心,兩對對映體,這對催化體系提出了更高的要求。但是,該催化體系卻顯示出了高度的兼容性。當使用二級烷基溴代物進行反應時,該反應可以一步構建兩個相鄰的手性中心,并以中等到優秀的產率,高對映選擇性和非對映選擇性得到手性產物(10a-10m)。尤為重要的是對于具有連續四個手性中心的手性三元環結構,該反應依然可以取得優良的產率和高立體選擇性(10g),這是其他方法很難做到的,從而體現出該方法的優越性和高效性。▲圖3 圖片來源:J. Am. Chem. Soc.
接下來,作者考察了環丙基鋅試劑與一系列親電試劑的EDRC反應(圖4)。在完成了第一步鎳催化下立體選擇性偶聯反應之后,作者對不同第二組分親電試劑進行了考察。烯丙基溴,酰氯和炔基溴代物等親電試劑均能與剩余的另一構型鋅試劑發生立體專一性偶聯反應,得到不同取代的、構型相反的手性三元環產物(5b-5f)。同時,該立體專一性反應也適用于其他親電試劑,如NBS、NCS以及碘,生成手性三元環鹵代物(5g-5j)。需要強調的是這些手性三元環鹵代物是重要的手性合成子,在有機合成中具有重要應用。此外,硒或者錫取代的手性三元環(5k, 5l)也可以通過該策略得到,從而進一步體現了該EDRC反應的實用性。▲圖4 圖片來源:J. Am. Chem. Soc.
該反應的實用價值還體現在產物的多樣性轉化上。以EDRC得到的兩種構型的產物13和5h為例(圖5):化合物13的炔基官能團,不僅可以進行Sonogashira偶聯反應向分子中引入芳雜環,還可以轉化為α,β-不飽和酮或者通過[3+2]反應生成一系列雜環等。同時,由于二氟亞甲基對炔基的活化作用,無銅催化的Click反應也能高效進行進行。手性三元環化合物5h的重要性同樣可以通過一系列轉化得到體現。5h可以通過鋰溴或者鎂溴交換生成環丙基金屬試劑,其與一系列的親電試劑發生立體專一性的反應,可以制備手性膦配體、三元環硼酸酯和硅類化合物以及手性三元環羧酸、酰胺、腈類化合物,從而體現了該手性發散式合成的優越性。▲圖5 圖片來源:J. Am. Chem. Soc.
張新剛課題組首次提出了對映發散式接力偶聯(Enantiodivergent Relay Coupling, EDRC)的策略,并將該策略應用于鎳催化的消旋環丙基鋅試劑與1級、2級(氟)烷基鹵代物的不對稱接力偶聯反應中,實現了一鍋法合成具有不同取代,構型相反的的手性三元環化合物,為過渡金屬催化的不對稱偶聯反應提供了新思路。該工作發表于J. Am. Chem. Soc. DOI: 10.1021/jacs.0c04462,其中安倫博士和童非非為共同第一作者。在該文的投稿過程中,加州理工學院的Gregory C. Fu教授發表了一篇有關鎳催化下消旋親核試劑與1級,2級消旋親電試劑的偶聯反應(Science 2020, 367, 559–564)。該工作得到了國家自然科學基金和中國科學院先導專項B的資助。張新剛課題組隸屬中國科學院上海有機化學研究所,中科院有機氟化學重點實驗室。課題組主要從事有機氟化學研究,目的是通過新催化體系的建立,對我國廉價含氟資源和工業廢氣物進行高效轉化,解決其中存在的重要科學問題,發現有機氟化學新反應,發展新概念和新方法,并進而實現含氟分子的功能化,合成特種含氟功能材料和具有生物活性的含氟分子。具體研究方向:1)基于廉價含氟資源的高效轉化,包括廉價金屬催化的氟化、氟烷基化、氟芳基化以及不對稱反應研究;2)金屬二氟卡賓化學;3)生物大分子選擇性氟修飾及其在化學生物學中的相關研究。