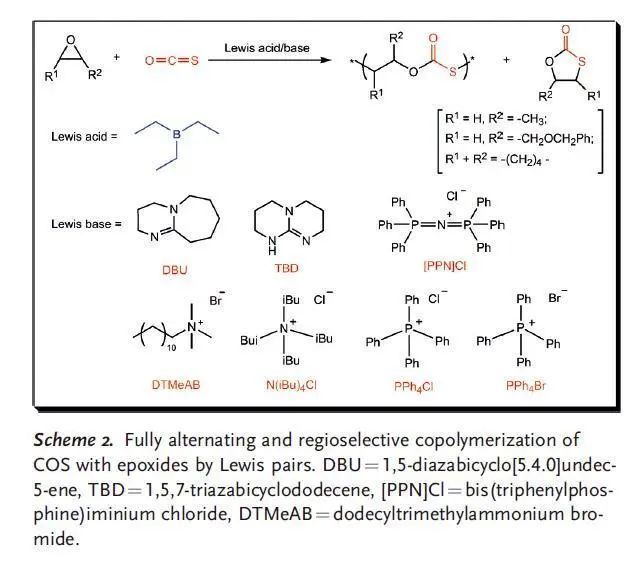
給大家分享一篇2015年發表在Angewandte Chemie International Edition上的文章,題為:Perfectly Alternating and Regioselective Copolymerization of Carbonyl Sulfide and Epoxides by Metal-Free Lewis Pairs,在本研究中作者利用脒,胍和季鎓鹽作為Lewis堿;三乙基硼烷(TEB)作為Lewis酸組成無金屬Lewis酸堿對,催化羰基硫(COS)和環氧烷烴完全交替和區域選擇性共聚,成功地獲得了高透明性的聚(單硫代碳酸酯),其尾-頭連接含量超過99%,分子量高達92.5 kg/mol。在大多數情況下O/S交換被有效地抑制,在室溫下TOF值可達119 h-1。本文的通訊作者是浙江大學的張興宏教授。
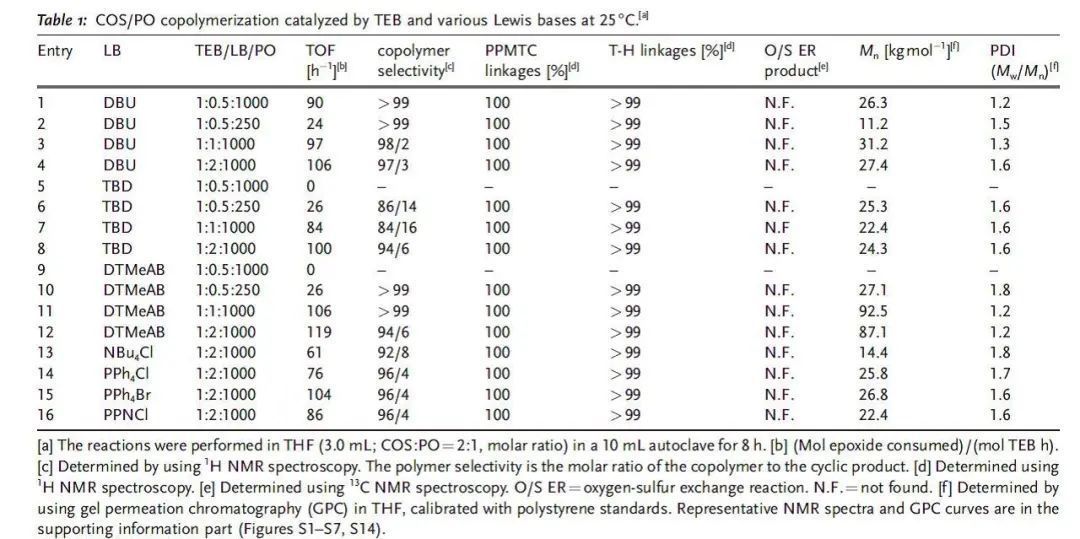
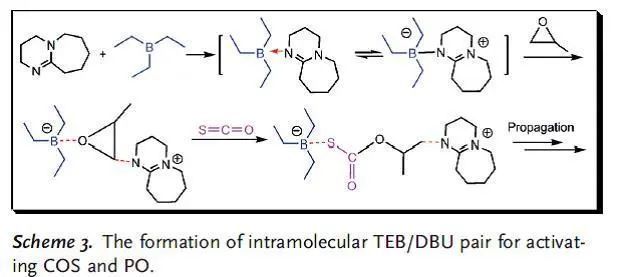
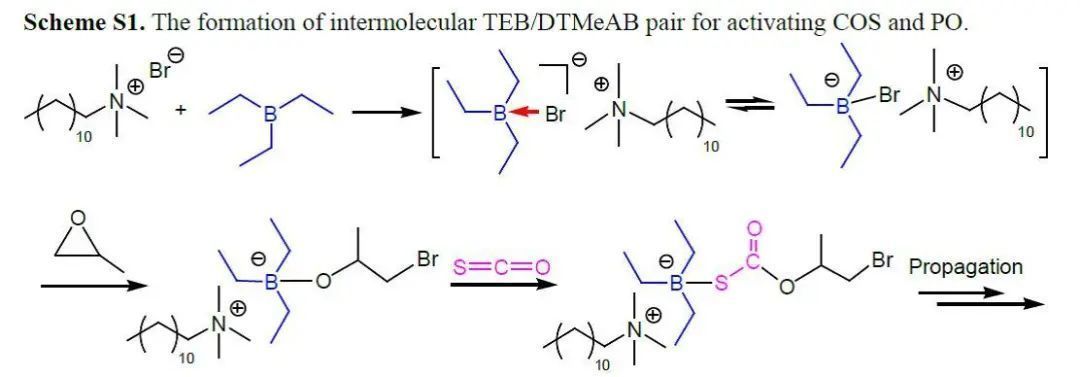
對于涉及CO2,COS或CS2的共聚過程,配體穩定的金屬催化劑通過配位插入機理進行反應,其中金屬配合物作為Lewis酸中心活化環氧烷烴,季銨鹽和有機堿作為Lewis堿,對于提高金屬催化劑的催化活性是必需的。這些金屬Lewis酸/有機堿體系的催化活性和選擇性可以通過調節金屬配合物的配體結構來巧妙地調節。最近,已成功開發出無金屬的Lewis酸堿對催化乙烯基和雜環單體的聚合。與傳統的金屬/堿催化劑體系相似,這些Lewis酸堿對之間存在配位鍵,可插入小分子。 作者設想有機Lewis酸堿對可以代替COS與環氧烷烴共聚過程中的典型金屬陽離子/有機堿,與金屬催化過程相似,Lewis酸堿對與環氧烷烴綁定后開環,隨后交替插入COS和環氧烷烴形成共聚物。Lewis酸的酸性,Lewis堿的親核性和Lewis酸堿對的立體效應是影響無金屬催化劑有效性的決定因素。廣泛應用的Lewis酸堿對如B(C6H5)3/PPh3,B(C6F5)3/PPh3,B(C6H5)3/DBU(TBD),和B(C6F5)3/DBU(TBD)一開始被用于催化COS與PO共聚,但在25 ℃和80 ℃下均無反應。Gnanou與Feng等發現,鹵化鎓或醇鹽和三乙基硼烷(TEB)可以成功地催化CO2與PO或CHO共聚反應。基于這項研究工作,作者開發了一種完全交替且完美區域選擇性的COS/環氧烷烴共聚方法,TEB與各種Lewis堿(包括脒,奎尼丁和季鎓鹽)組成Lewis酸堿對作為催化劑,得到無色且高度透明且結構明確的聚(單硫代碳酸酯)(圖1)。 圖1 與金屬催化體系相同,類似的COS共聚過程有兩個特殊的問題。一個是COS與環氧烷烴間的O/S交換反應導致在主鏈中產生混合的重復單元,另一個是COS與單取代環氧烷烴獨特的立體化學控制。 表1 表1總結了反應8小時在不同TEB/LB/PO投料比下,TEB與多種Lewis堿催化的COS/PO共聚反應。得到的聚合物從乙醇中沉淀后,可以通過熱壓方法將白色純產品制成透明晶片。通過1H NMR顯示所有聚合物都具有完全交替的結構,這說明COS的插入比連續的PO均聚更快。13C NMR顯示所有聚合物具有超過了99%的尾-頭連接含量,優于(salen)CrX催化體系。這符合之前提出的機理,鏈增長的活性位點選擇性地連接PO上空間位阻較小的亞甲基碳。此外,在所有聚合物的1H和13C NMR中沒有觀察到O/S交換,因此,這些Lewis酸堿對催化劑提供了完全交替的區域選擇性的COS/PO共聚物。需要特別指出的是,Lewis堿的性質與TEB/LB/PO的投料比對反應的活性、聚合物的選擇性和分子量有很大的影響。當TEB/DBU/PO = 1.0/0.5/1000時(entry 1),TOF值為90 h-1,選擇性 >99%,得到聚合物的分子量26.3 kg/mol,分子量分布1.2。當減少PO的量后(entry 2),TOF值和分子量分別減少到24 h-1和11.2 kg/mol。增加DBU的量可以提高TOF值(97 h-1和106 h-1)和Mn,而共聚物的選擇性僅稍有犧牲(entry 3和4)。在用TBD作為Lewis堿代替DBU時,催化活性的波動是相似的,但值得注意的是,當TEB/TBD/PO = 1.0/0.5/1000時,8小時內沒有得到聚合物(entry 5),這可能是PO的高負載削弱了Lewis酸堿對之間的配位鍵。同時,高負載的TBD導致聚合物的選擇性下降,原因可能是TBD的堿性稍強。相比之下,較弱的堿DMAP與TEB結合使用時,在相同的反應條件或更高的溫度下,無法催化COS/PO共聚反應。 一些季鎓鹽在與TEB配對時也可以有效地催化COS/PO的共聚反應。與TBD相似,DTMeAB在低濃度下無法催化反應(entry 9);在投料比TEB/DTMeAB/PO = 1.0/1.0/1000時是有效的(entry 11);繼續增加DTMeAB的量導致催化活性略有提高,但聚合物的選擇性降低。此外,就催化活性和聚合物選擇性而言,其他季銨鹽(如NBu4Cl,PPh4Br和PPNCl)與DTMeAB相似。大體積陽離子NBu4Cl和PPh4Cl(entry 13和14)連同強親核性陰離子Br-和Cl-(entry 15和16)趨向于增加反應的TOF值。通常,較少的Lewis堿和較大的PO負載量有利于形成具有窄PDI的聚合物。特別的是TEB/DTMeAB組成的Lewis酸堿對可以非常有效地得到窄PDI(1.2)和高分子量(87.1-92.5 kg/mol)的聚合物。聚合物的GPC通常呈現雙峰,原因是微量的水導致的鏈轉移。 作者還研究了溫度對催化反應的影響。分別選擇TEB/DBU與TEB/DTMeAB體系,研究發現提高反應溫度或增加Lewis堿的用量對聚合物的選擇性有非常不利的影響。在高反應溫度和高TEB負載下也觀察到O/S交換產物。實際上,對于在70-80 ℃溫度下TEB/DBU催化的共聚反應,產物100%為兩種環狀硫代碳酸酯。 為了更深入地理解引發步驟,通過1H NMR對一系列Lewis酸堿對和單體進行了表征。在核磁上可以觀察到TEB/DBU Lewis酸堿對的形成,再加入1當量的TEB后,DBU上Ha的化學位移從2.32 ppm移動至2.59 ppm。這種去屏蔽作用表明DBU的氮原子與硼烷中心之間形成配位鍵。當加入1當量PO后,觀察到Ha的化學位移從2.59 ppm移動至2.63 ppm,說明存在進一步去屏蔽作用。同時PO的1H化學位移從2.99, 2.76, 2.44 ppm相應地移動到2.82, 2.58, 2.27 ppm。這一觀察表明PO被Lewis酸堿對活化。相反,向TEB/DBU Lewis酸堿對中添加過量的COS會導致DBU上Ha的化學位移向高場移動至2.45 ppm,這表明形成了較不穩定的DBU-COS-TEB中間體。將PO和過量的COS同時添加到TEB/DBU Lewis酸堿對中后,DBU上Ha化學位移向低場移動至2.69 ppm,更接近DBU-PO-TEB系統的化學位移。這種去屏蔽作用的增強表明PO可能在DBU-PO-COS-TEB加合物中開環,并充當了鏈增長物種(圖2)。 圖2 相比之下,TEB與DTMeAB的Lewis酸堿對中DTMeAB NCH3上的質子(Hb)表現出明顯的屏蔽作用,這說明[N(CH3)3C12H25]+Br-比[TEB-Br]-[N(CH3)3C12H25]+離子對作用更強。通過對1H NMR的比較發現,向TEB/DTMeAB Lewis酸堿對中引入PO或COS后對Hb的化學位移的影響可以忽略不計,但PO的1H化學位移從2.99, 2.76, 2.44 ppm相應地移動到2.78, 2.54, 2.22 ppm,表明DTMeAB對PO有明顯的活化作用。當同時向TEB/DTMeAB Lewis酸堿對中加入等當量的PO和過量的COS時,Hb的質子僅向高場輕微移動(3.27至3.25 ppm)。同時,PO的1H化學位移表明發生了開環。提出了一種可能的中間體[TEB-COS-PO-Br]-[N-(CH3)3C12H25]+(圖3)。由TEB/DTMeAB催化的COS/PO共聚過程的誘導期約為1-2小時,表明這種引發物質的形成是速率控制步驟。 圖3 作者認為與環氧烷烴具有配位鍵的四面體硼烷陰離子部分是共聚反應的引發劑,這一物種通過大體積陽離子(圖2中的分子內陽離子和圖3中的分子間陽離子)被穩定。值得注意的是,在相同反應條件下用較大的(C6F5)3B或(C6H5)3B取代TEB時,未發生聚合反應,說明適當的酸度/堿度和Lewis酸堿對的大小對于COS/PO共聚反應是必要的。 使用這種Lewis酸堿對催化劑其他環氧烷烴與COS也可以實現共聚反應。值得注意的是,CHO與COS共聚時抑制了環狀碳酸酯的產生,這是由于其能壘比其他環氧化物要高。苯基縮水甘油醚(PGE)和COS在TEB/DTMeAB和TEB/DBU的催化下也可以發生共聚,所得聚合物完全交替且尾-頭連接含量 >99%。但反應的TOF值較低,并且在TEB/DBU催化時產物90%為環狀硫代碳酸酯。此外,這些Lewis酸堿對在使含有吸電子取代基的環氧烷烴(如氧化苯乙烯)與COS共聚時完全無效。 在本文中,作者通過使用無金屬的Lewis酸堿對合成了完全交替且高選擇性的聚(單硫代碳酸酯),其尾-頭連接含量超過99%,分子量高達92.5 kg/mol。在大多數情況下O/S交換被有效地抑制,在室溫下TOF值可達119 h-1。接下來的工作是進一步研究催化過程的機理,并開發出更具活性的Lewis酸堿對,以實現多種環氧烷烴的共聚。 DOI: 10.1002/anie.201701780 文章及圖片來源: https://onlinelibrary.wiley.com/doi/abs/10.1002/anie.201701780