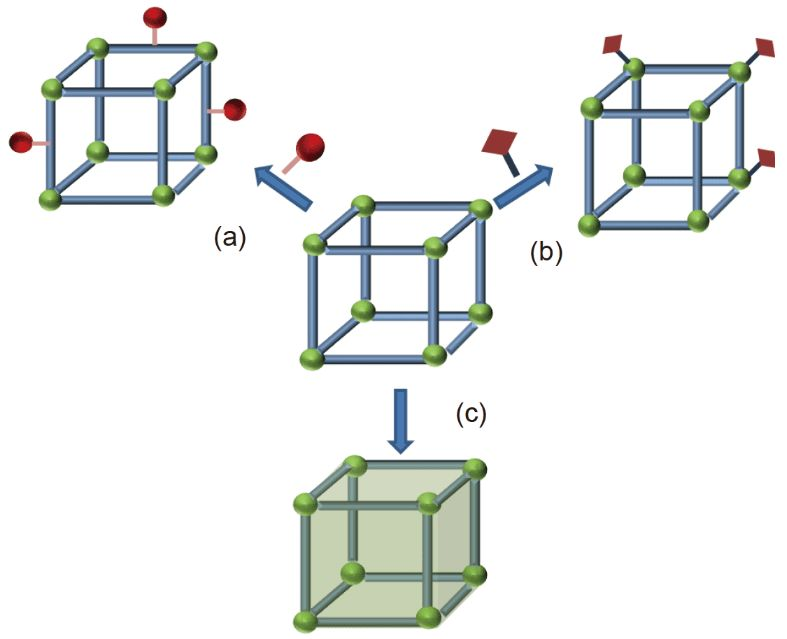
電化學CO2還原反應(electrochemical CO2 reduction reaction eCO?RR)可將CO?轉化為高附加值化學品,是一個存儲與利用可再生能源的理想策略,對實現碳中和目標與可持續發展具有重要意義。近年來,氮摻雜碳基底負載的鐵單原子催化劑(Fe-N-C)因其低廉的成本與優異的催化性能成為備受矚目的eCO?RR催化劑,實驗表明其高活性位點是三價鐵(Fe(III))中心,而二價鐵(Fe(II))中心的催化活性較低。理論研究通常采用FeN?-C模型來模擬Fe-N-C,但其中的Fe具有兩個未配對電子,是典型的Fe(II) 中心,因此無法解釋Fe(III)-N-C優異的催化性能。 清華大學肖海團隊采用高精度波函數方法校準的DFT+U計算與巨正則系綜模擬,確定了Fe(III)-N-C的理論模型,并由此揭示了Fe(III)-N-C單原子催化劑在eCO?RR工況下經歷電勢依賴的活性位點結構演化,“放飛自我”自發形成了一類瞬態的懸掛活性位點(如下圖中的紅色結構所示),具有催化eCO?RR的高活性與高選擇性,闡明了Fe(III)-N-C優異的催化性能。該工作也表明了電化學氛圍驅動產生的高能亞穩態結構可能才是電催化構效關系的結構基礎。
以具有明確氧化態Fe中心的配合物分子作為參考(如下圖所示),研究團隊通過對比它們與各種Fe-N-C周期性模型中Fe的2p芯能級與自旋態,確定了:傳統的FeN4-C模型中Fe為二價,是Fe(II)-N-C單原子催化劑的理論模型;而FeN1C3-C模型中的Fe才是三價,是Fe(III)-N-C單原子催化劑的理論模型。 基于巨正則自由能的Pourbaix相圖表明(如下圖所示):在eCO?RR的電化學還原性氛圍下,Fe(II)N4-C的活性位點配位結構保持不變,而Fe(III)N1C3-C的活性位點配位結構則由于配位C原子的還原氫化發生了本質上的變化,其中所有的Fe-C鍵都被打斷,形成了僅保留單個Fe-N鍵錨定的懸掛活性位點,其中的Fe再由來自電解質的水分子飽和配位。從頭算分子動力學(AIMD)模擬也驗證了該結構在溶劑中的動態穩定性。 進一步的理論研究表明,這一類懸掛活性位點憑借其結構柔性,可以通過低能壘路徑高效活化CO?分子,并形成多路徑協同的反應網絡(如下圖所示);結合微觀動力學模擬所預測的Fe(III)-N-C催化性能與實驗吻合,從而在原子尺度上闡明了Fe(III)-N-C優異催化性能的構效關系。該發現為理性設計高性能eCO?RR單原子催化劑提供了新的理論認識與思路。



論文信息 Transient Dangling Active Sites of Fe(III)?N?C Single-Atom Catalyst for Efficient Electrochemical CO2 Reduction Reaction Yun-Ze Qiu, Xiao-Meng Liu, Wenying Li, Jun Li, Prof. Hai Xiao 文章的第一作者是清華大學化學系的博士生邱昀澤。 Angewandte Chemie International Edition DOI: 10.1002/anie.202424150