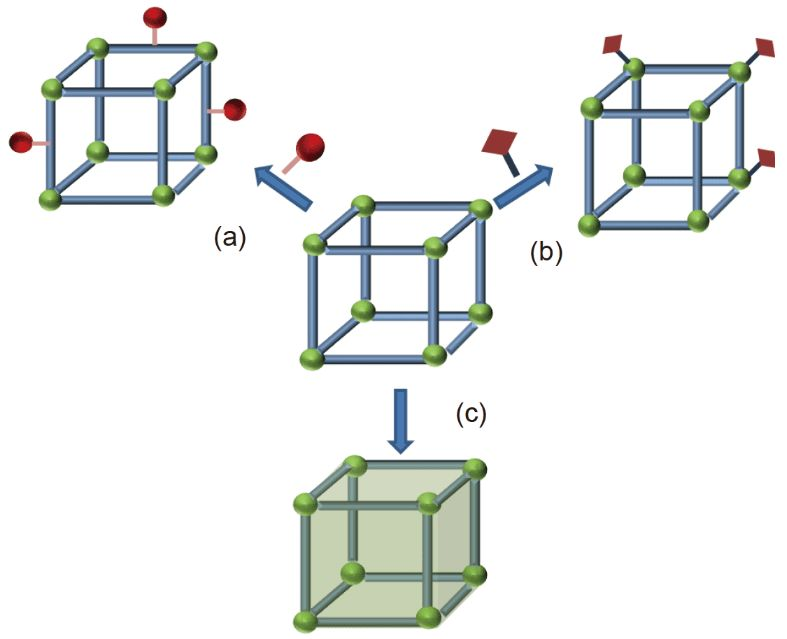
用[NHC-Ni(allyl)]BArF催化首次觀察到具有典型氫烷基化反應活性的亞甲基環丙烷(1)和未活化烯烴(2)的環化反應 (NHC = N-heterocyclic carbene)。結果表明,1的C-C裂解不涉及Ni0氧化加成,這在以前的體系中是至關重要的。因此,本文所報道的方法是實現亞甲基環戊烷(3)具有廣泛范圍的高度化合性和區域選擇性合成論文的一種補充。有效NHC/NiII催化1的重排導致在2存在下3的匯聚合成。
亞甲基環丙烷(1:MCP)是有機合成中的一個重要組成部分,近年來新的過渡金屬催化劑在其活化方面取得了重大進展 (Scheme 1)。除了使用傳統的方法,如光活化和熱重排,一些專門的MCPs,催化劑如 P/Pd0 和Ni0(AN)2(AN = Acrylonitrile),在1(近端或遠端位置)的氧化開環反應中特別有效,從而與烯烴(2)形成有效的C-C鍵。這一策略是獲得功能化環戊烷的最重要途徑之一 (3; Scheme 1 a)。然而,只有電子貧乏或小烯烴是分子間反應的有效伙伴。未活化或體積較大的烯烴即使在高溫下使用較長的反應時間也往往失效,這已被認為是實現更高選擇性和產品結構多樣性的關鍵障礙。插入金屬氫化物或其等價物代表另一種常見的MCP激活策略 (Scheme 1 b)。中間體,如環丙基甲基-Ni/PdII,經常進行β碳消除或其他合作伙伴的插入,為構建氫功能化產品提供了一條通用路線。最近,[NHC-Ni(allyl)]BArF (NHC = N-heterocyclic carbene)被認為是一種通過環丙烷和烯烴的交叉氫烷基化催化劑,它涉及環丙烷開環。因此,我們對用于MCP激活的[NHC-Ni(allyl)]BArF感興趣。
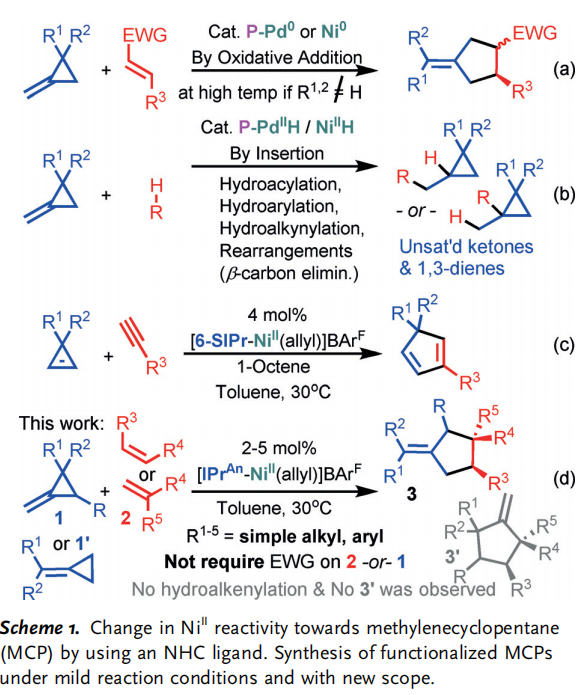
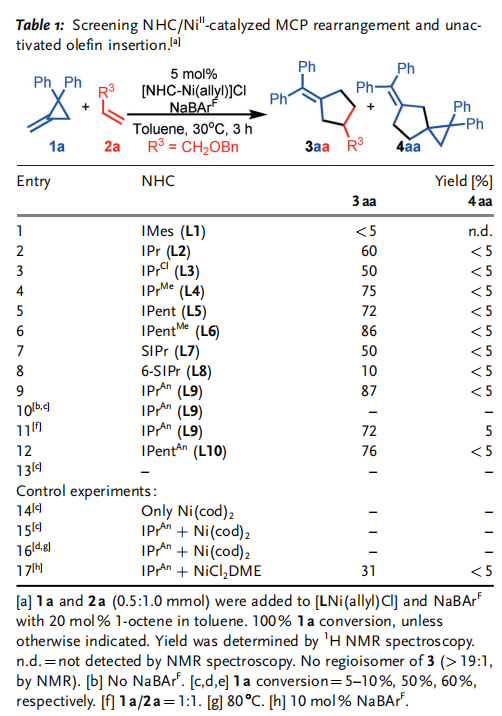
在此,首先使用[NHC-Ni(allyl)]BArF催化劑實現了1和2的環化反應(Scheme 1 d)。它提供了一種新的、高效的合成高價值和功能化的亞甲基環戊烷3的途徑。與幾種高度競爭的交叉氫烷基化反應形成鮮明對比。使用Pd0或Ni0之前沒有被證明的1和2的幾種組合(Scheme1a)現在是兼容的并且在環境條件下反應。并發癥,如不希望消耗1和2的非選擇性的同質反應/低聚,如許多相關的氫化物系統,和/或快速異構化被抑制。它避免了競爭的MCP開環[2+2]或[3+3],或將MCP插入Ni0,以及在使用AuI時重排成環丁烯等問題,所有這些都與其他的MCP反應有關。值得注意的是,對照實驗表明,3的形成不是NHC加速Ni0氧化加成1的結果,這一觀察結果與用NHC代替P所假定的反應活性明顯不同。他們的結果表明,NHC/NiII催化1的有效重排,從而達到了無可比擬的反應性、新的范圍、高選擇性和效率。作者的研究從一個簡單的MCP,1a和末端烯烴2a開始,使用[NHC-Ni(allyl)]BArF催化劑報道的環異構化和氫烷基化(Table 1, entry 2)。令人驚訝的是,沒有觀察到假定的交叉氫烷基化或β-碳消除產物,但首次觀察到一種新的[3+2]產物(3aa)。篩選NHCs表明NHCs在這種催化作用中起著重要的作用,使用空間體積大的NHC對所需的反應性至關重要(Figure 1; Table 1,entries 1–9, 12–13)。然而,NHC空間效應對高化學-(3aa/4aa)和區域選擇性(沒有其他區域異構體的核磁共振波譜)的影響尚不清楚。在那些結構多樣化,提供了所需的反應性的NHCs中,沒有發現明顯的趨勢。首先,假設3的形成是典型的MCP激活Ni0的結果,無論是從Ni0殘基還是在催化劑合成過程中意外的原位NiII還原。然而,使用Ni0的對照實驗表明,在溫和的反應條件下,不能形成3aa (entry 14)。此外,盡管對結構優化的NHCL9進行了鑒定,但在相同的反應條件下,相應的Ni0/L9即使在80℃下也沒有期望的反應活性 (entries 9, 15, and 16)。在沒有NHC配體或BArF作為陰離子的情況下,觀察到1a和2a的低或非選擇性轉化(entries 10 and 13)。此外,L9/NiCl2的表現甚至優于L9/Ni00 (entry 17)。值得注意的是,這里沒有觀察到3’aa(Scheme 1d)。這是在環丙烷(CP)環和丙烯酸酯上存在不平衡的MCPs的相關Ni0體系中注意到的主要異構體(Table 2)。總之,從Ni0獲得的上述結果表明,3aa在這里的形成不太可能是NHC輔助Ni0氧化加成MCP的結果 (Scheme 1a)。最后,應該指出,陰離子對反應活性有顯著的影響(Table 1, entries 9 and 10; BArF vs.Cl)。
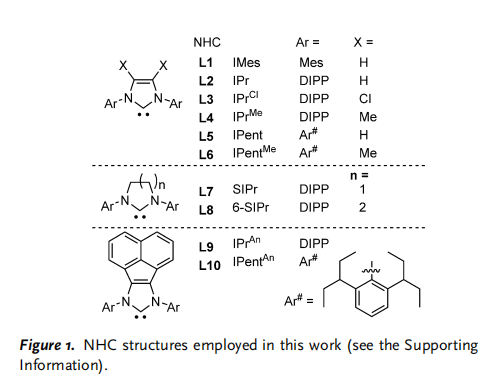
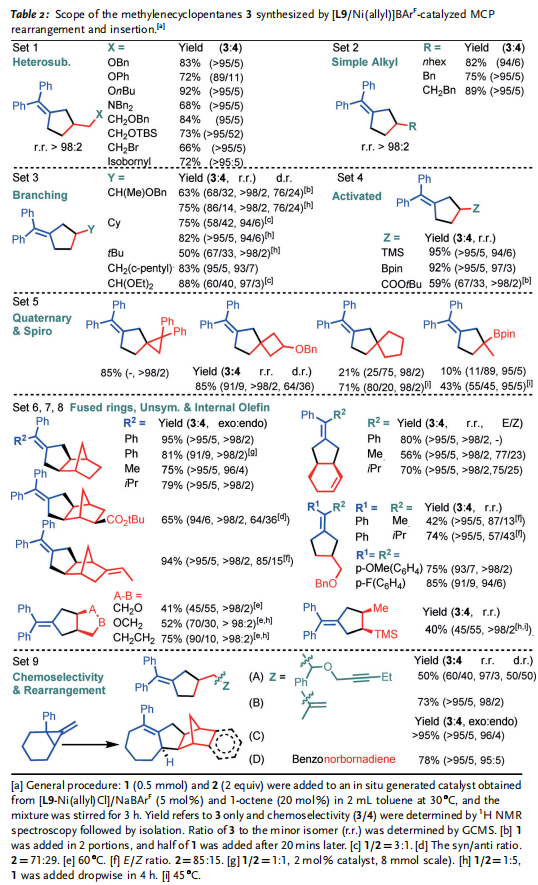
新的底物范圍和的和無與倫比的反應性進一步支持了典型的Ni0催化3的選擇性合成(Table 2)。他們的催化在低催化劑負載和環境條件下有效地進行。雜原子取代的末端烯烴(X = 醚、胺和烷基鹵化物; Table 2; Set 1),分支部分 (Y= 烷基和雜原子取代的三元無環/環,四元無環,和β-分支; Set 3),以及非常簡單的烷基(R = 線性烷基、芐基和同芐基; Set 4),現在作為相容的底物,而不僅僅是乙烯,以及其他體系中通常需要的缺乏電子和活化的烯烴(Z=ester)[Set 4; c.f. 3’via Ni(AN)2],在這里值得注意的是,第一次成功地使用乙烯基-TMS和乙烯基Bpin作為2,因為它們是有用的后改性處理,以制造其他甲基環戊烷。穩健的反應條件鼓勵作者他們探索更大的底物(2)和不同的取代1(Table 2; Set 5-8),值得高興的是,以前沒有建立的幾種替代模式現在被廣泛接受,提供了一種新的直接途徑來訪問更復雜的結構。底物如gem-olefins被確定為MCPs的新合作伙伴,導致產品具有季碳中心和螺旋結構(4 aa; Set5)。事實上,主要是當使用不匹配的2時,次要產品通常被確定為4aa。這一結果表明,我們反應的高化學選擇性(3/4)是基于底物對的選擇和低的1/2比,通過Table 1中 Sets 1-4與Set5和entries 9和 11的結果可以看出。這里報道的微調催化劑反應性也與具有其他取代基的1兼容,而且,它也很好地適用于一系列對稱和不對稱的內部烯烴 (Table 2; Sets 6-8)。新大陸的發現是十分有作用的(as low as 2 mol% cat., 1/2 = 1:1)直接提供具有高外選擇性的熔融環結構。MCP取代基的選擇可以是混合芳烴,也可以是脂肪烴,具有較大的R1和R2基的MCPs具有較高的反應性 (e.g. Ar> iPr> Me),大概是因為一個更有效的開環輔助增加的空間排斥組間。具有電子不同取代基 [p-X(C6H4), X = H, OMe, F]的底物(1)的可比較產率在一定程度上支持了上述假設(Sets 1 and 7, 75-85%),在某些情況下,3的產率較低,這是由于相對于homo-MCP(4)形成的反應性較低。然而,它們的產物區域選擇性仍然很高 (大部分情況中> 50:1 )。催化作用也是化學選擇性的 (Set 9)。具有端基而不是gem-olefin的底物2是合成1的首選反應物。令人驚訝的是,含有內部炔的底物在這一反應中保持完整,盡管它在典型的M-H插入反應中具有反應性,因此,與我們以前的系統的反應性形成鮮明對比。最后,使用帶有熔融環的MCP產生了環膨脹重排,代表了一種輕松組裝復雜的熔融環結構的方法。

作者測試了1’a和1a的合成 (Scheme 2b)。希望能提供見解以及合成的替代品。事實上,它按預期進行[vs.Ni0],表明1a和1’a共享一個公共中間體。一個較慢的反應被注意到1’a,這是一致的空間關注在這些交換在Ni0氧化加成。用二烷基取代的1的合成有時是困難的,然而Scheme 2c中反應的成功提供了一種很好的選擇來合成二烷基結構。典型的臭氧溶解給我們環戊酮的產率很好(Scheme 2d)。
在此階段,由于NHC/Ni0被驗證為他們所需反應的非活性物種,因此快速合成具有高化學和區域選擇性和廣泛范圍的3的關鍵是主要NHC/NiII的研究(Scheme 2)。確切的機制仍在研究中,但考慮了烯丙基交換途徑,因為沒有觀察到1和2的交叉氫烷基,并且由于3不能從插入1和β碳消除獲得。因為i)在 [L-NiII-(allyl)]BArF上烯丙基的交換可以在室溫下進行,ii)已知MCP重新排列并賦予烯丙基兩性物種(熱條件下的二氧基MCP)。這里可能的途徑之一可能與L9/NiII催化的Int.-A的形成有關(Scheme 2a)。它可能是由開始時與催化劑的烯丙基-烯丙基交換形成的,也可能是插入2后的烷基-烯丙基交換形成的。

總之,利用NHC改變NiII催化劑對1的反應活性,建立了1(或1’)和2給3的高效環化。觀察到高效環化對氫烷基化反應活性,并歸因于有效和選擇性重排1,而不是烯烴插入時,使用[L9/NiII(allyl)]BArF。NHC/NiII體系與典型的Ni0/Pd0體系在范圍和反應性方面具有很高的互補性。第一次,簡單的未活化烯烴,而不是電子貧乏的不飽和羰基底物,是取代1的良好伙伴,即使在環境條件下也是如此。此外,NHC/NiII催化劑獲得了較高的3~3’比,而Ni0催化劑通常相反。總的來說,在這里發現的1和1’的溫和激活途徑為在以前的Ni0條件下實現較高的化療和區域選擇性提供了新的基礎。最后,本報告提供了一種簡單的方法來制造有用的環戊烷,以及來自簡單烯烴的螺旋和熔融環系統,這些環系統很難直接從簡單的化學原料中獲得,并且具有如此廣泛的襯底范圍。
Angew. Chem. Int. Ed., 2019, 58, 5702-5706, DOI: 10.1002/anie.201901255